Day 1 wrap up
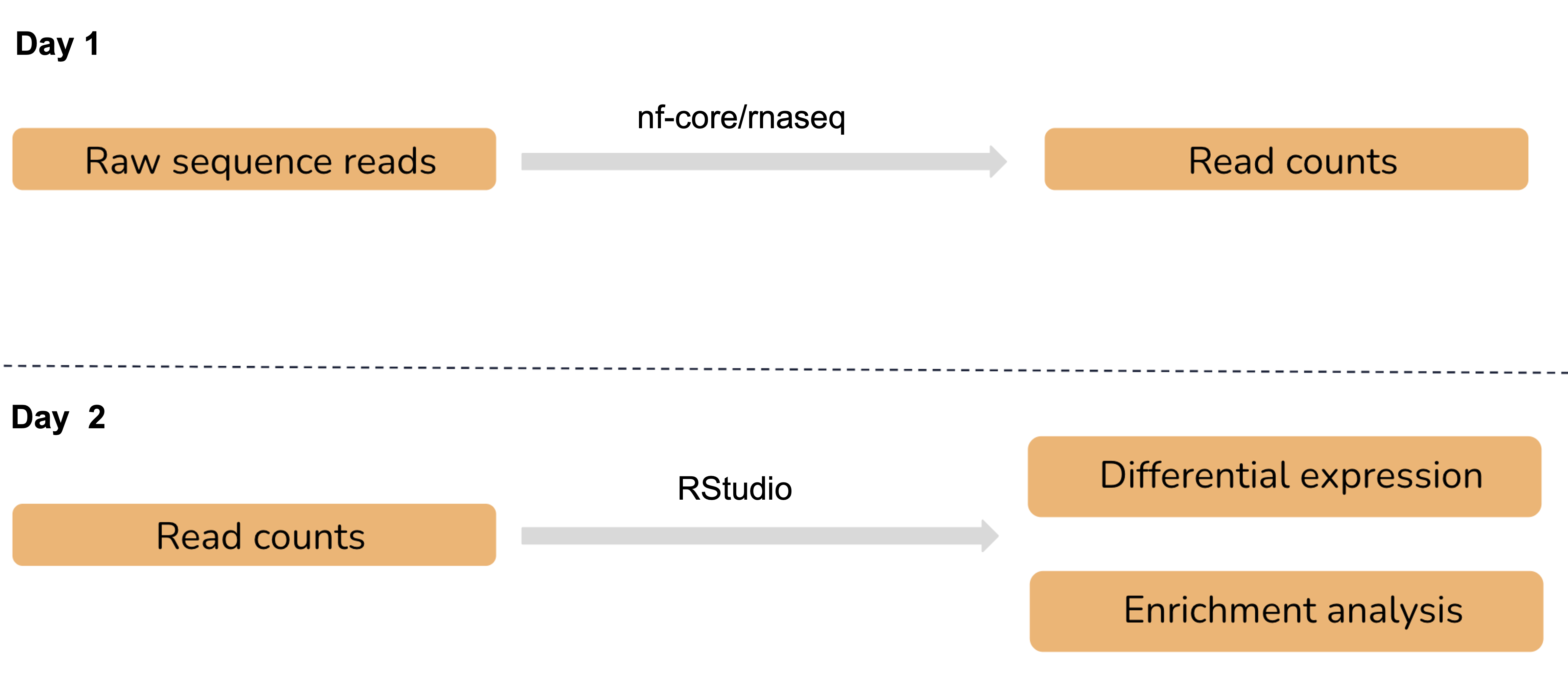
In this session, we performed the necessary data pre-processing steps required for differential expression with RNAseq data. We used the nf-core/rnaseq pipeline to process raw data from fastq inputs, align sequence reads, generate gene counts and perform extensive quality control. This pipeline is built using Nextflow, which is a bioinformatics workflow management tool that supports reproducible, portable, and scalable analyses. Tomorrow, we will use the count matrix generated by this pipeline to identify differentially expressed genes and perform functional enrichment analysis. We will be working interactively with our data in RStudio.
Key takeaways for day 1
- Raw data quality control is an essential step, it allows you to identify any potential issues that may interfere with analyses.
- The nf-core/rnaseq pipeline follows community best practices and offers a reproducible, portable, and user friendly method for pre-processing your RNAseq data.
- FastQC is a useful tool for fastq quality inspection, however it was not built for RNAseq data so your RNAseq data will always fail some of their tests.
- Read trimming is not always necessary. Your choice to trim reads will depend on their quality, presence of adapter sequences, and read alignment tool of choice. Over-trimming or unnecessary trimming can sometimes remove valuable sequence information, so it’s essential to strike a balance.
- RNAseq read alignment is splice-aware, meaning that it’s designed to identify and handle reads that span exon-exon junctions. This capability ensures that such reads are accurately mapped, providing a true representation of the transcriptome.
- Read quantification to create a count matrix for your samples is required for differential expression analysis. Proper quantification will ensure that RNA abundance is accurately represented.
- Different alignment and quantification tools and methods suit different applications. These tools will have different underlying models and assumptions that will be reflected in their outputs.