Data pre-processing with nf-core/rnaseq
- Understand how to apply parameters to an nf-core pipeline to cusomise its execution
- Understand the nf-core workflow execution standard output
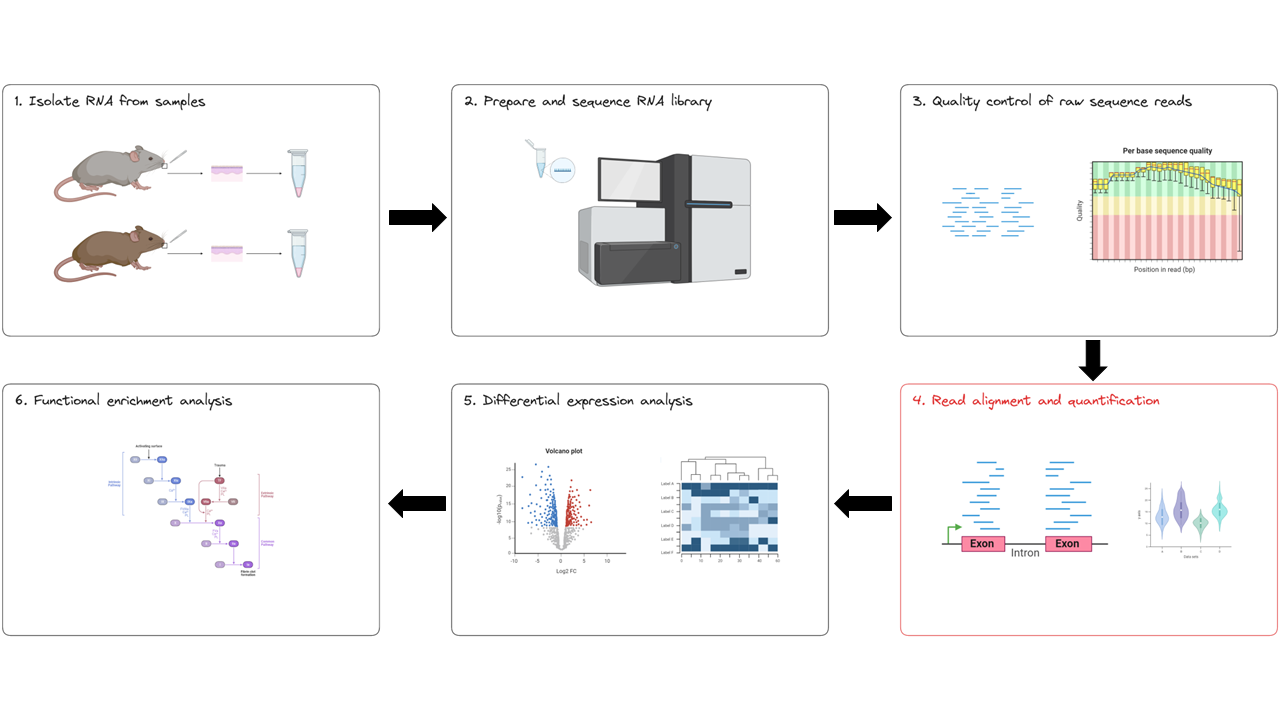
Now that we have confirmed our data is of good quality and trimmed low quality bases and adapter sequences from our reads, we can proceed with running the remainder of the nf-core/rnaseq pipeline to complete the read alignment and quantification steps (red box below).
 .
.
1.3.1 Run the data preprocessing workflow
A nice feature of Nextflow and nf-core pipelines is the ability to resume an incomplete workflow execution from the last successfully completed step. We will use this feature to save ourselves time and pick up from after read trimming.
One of Nextflow’s key features is the ability to resume a workflow from the point of failure, rather than starting over from scratch. When running any Nextflow workflow, you can use the -resume flag to use this feature.
Nextflow uses a caching system to track the results of each task in a workflow. This cache will include both the input files and output files created for each task. If your workflow execution is interrupted for any reason, some tasks may have completed while others have not. Using -resume tells Nextflow to check the cache to see which tasks have already finishe. Notice the appearance of a work directory when you run your Nextflow pipelines. For tasks that were not completed or quit with an error, it will attempt to rerun them.
➤ In your terminal, check where you are by running:
pwd/home/training/Day-1If you are not in the Day-1 directory, move into it by running:
cd /home/training/Day-1 We are going to adjust our run command slightly from last time, note:
- The
-resumeflag at the end of the command - We will output our results in a different directory (
--outdir) - We have added an extra flag to skip duplicate marking (
--skip_markduplicates)
Note that we have not specified a read alignment or quantification tool. We will be running the default setting which runs STAR to map the raw fastq reads to the reference genome and perform the read quantification using the alignment files with Salmon. You can specify which alignment method you’d like to use with --aligner.
By default, the nf-core/rnaseq pipeline uses picard to mark the duplicate reads identified amongst the alignments to allow you to guage the overall level of duplication in your samples. Unless you are using unique molecular identifiers (UMIs) it is not possible to establish whether your duplicate sequence reads are true biological duplication or PCR bias introduced during the library preparation.
Given our data hasn’t been tagged with UMIs, we won’t be able to accurately remove artifact duplicates from the alignments. Therefore we don’t need to worry about tagging them.
➤ Execute the following:
nextflow run nf-core-rnaseq_3.12.0/3_12_0/main.nf \
--input ~/Data/samplesheet.csv \
--outdir WBS-mouse-results \
--fasta ~/Data/mm10_reference/mm10_chr18.fa \
--gtf ~/Data/mm10_reference/mm10_chr18.gtf \
--star_index ~/Data/mm10_reference/STAR \
--salmon_index ~/Data/mm10_reference/salmon-index \
-profile singularity \
--skip_markduplicates \
--max_memory '6.GB' \
--max_cpus 2 -resumeToday we are using a pre-indexed reference genome (--star_index and --salmon_index) we prepared for you. Index files are required for tools like our aligners to run faster. Think of it like the index at the back of a textbook, where you can easily find what pages certain words are on.
Note that indexing requires a lot of memory and can be slow, so if you didn’t have a prepared index and you needed nf-core/rnaseq to perform indexing, you would need to increase memory (--max_memory).
The progress of the workflow will be displayed in the terminal and updated in real-time. Take a look at all the processes you have just run with a single command!
1.3.2 Customise the run command
The nf-core/rnaseq pipeline can be run using a single command with default parameters. The required parameters outlines a few basic requirements:
- An input sample sheet (
--input) - A reference genome, either one available through Illumina’s iGenomes database (
--genome), or a user-specified reference assembly (--fasta) and annotation file (--gtf) - A configuration profile suitable for the computing environment you’re working on (
-profile).
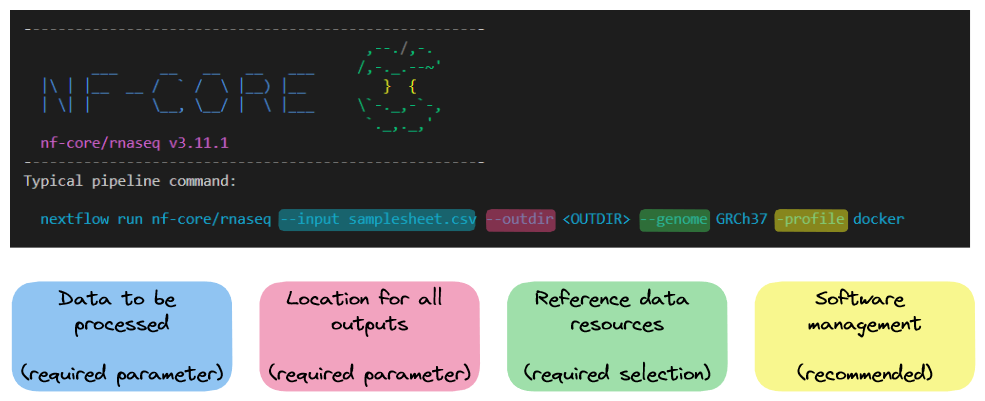
Compared with the run command we executed, a user working with human samples, who has Singularity installed and all fastq files stored in a directory would be able to run this command to run the nf-core/rnaseq pipeline:
nextflow run nf-core/rnaseq \
--input samplesheet.csv \ # Samples and their fqs
--genome GRCh38 \ # Illumina iGenomes database
-profile singularity # For pre-installed softwareMost of us will need to customise the command a little more than this though, depending on the resources available on our system, our dataset, and our analytical needs. Take a look at the nf-core/rnaseq usage instructions to determine what is right for you.
Nextflow allows us to set our configuration needs within a custom configuration file and apply it to our execution command using the -c flag.
Someone may have already built and shared a configuration file specifically for your infrastructure, like this config we built for Pawsey’s Nimbus instances. These pre-built, infrastructure specific configs are called Institutional configs and they can be downloaded with the pipeline code base and applied to all nf-core pipelines.
Institutional configs help us create efficient workflows that can be shared with others to reproducibly run the workflow in the same computational environment.
Once the pipeline has successfully completed, you will again see a message printed to your terminal that outlines how long it took your pipeline to run, how many tasks were executed, and how many of those tasks were cached:
-[nf-core/rnaseq] Pipeline completed successfully -
Completed at: 29-Sep-2023 03:55:58
Duration : 15m 12s
CPU hours : 0.4 (14.5% cached)
Succeeded : 153
Cached : 17➤ We will explore the outputs in depth in the next lessons. For now, list out the contents of your output directory:
ls -lh WBS-mouse-resultstotal 28K
drwxrwxr-x 7 training training 4.0K Sep 29 03:55 .
drwxrwxr-x 9 training training 4.0K Sep 29 03:40 ..
drwxrwxr-x 2 training training 4.0K Sep 29 03:41 fastqc
drwxrwxr-x 3 training training 4.0K Sep 29 03:55 multiqc
drwxrwxr-x 2 training training 4.0K Sep 29 03:55 pipeline_info
drwxrwxr-x 17 training training 4.0K Sep 29 03:55 star_salmon
drwxrwxr-x 3 training training 4.0K Sep 29 03:41 trimgalore- The nf-core/rnaseq pipeline offers users a high degree of flexibility and customisation whilst enhancing reproducibility.
- Users can modify parameters, customise configurations to tailor the pipeline’s execution to the needs of their compute environment, experimental design, and research objectives.
- Nextflow and nf-core pipelines employ a cache system for workflow execution, allowing users to resume workflows from any point of failure by using the
-resumeflag.