BioShell instances arrive with bioinformatics software and some reference data already installed. You don’t have to compile tools, manage dependencies, or track down container images before you can start.
What’s installed
Every instance comes with a core set of tools:
| Tool | Purpose |
|---|---|
| Python 3 | General-purpose scripting, and the language much of bioinformatics is built on |
| R | Statistical analysis and visualisation |
| JupyterLab | Browser-based notebooks holding code, plots, and notes in one place |
| RStudio | Browser-based development environment for R |
| Nextflow | Runs reproducible, scalable analysis pipelines |
| nf-core | Utilities and configurations for running nf-core pipelines |
| Globus Connect Personal | Make your VM a Globus end point for easy data movement |
Some of these are on your PATH and ready to type; others are modules you load first. To see
everything available on your instance:
module avail
Then load what you need, for example module load jupyter or module load rstudio. Anything
not listed, you can install yourself with Shelley.
How BioShell manages bioinformatics tools
Bioinformatics often requires us to use many different software including command-line software, R and Python packages. BioShell gives you access to over 100,000 bioinformatics packages, managed in three layers for you:
-
CernVM-FS is a read only filesystem that acts as a repository for 13,000+ tools and 118,000+
versions from BioContainers. It is available in your BioShell VM at
/cvmfs/. It looks like an ordinary folder, and files are fetched only when you use them, to help you manage your disk space. - sHPC packages containers in cvmfs into installable modules
-
Lmod is the system behind the
modulecommand you use to load and switch tools
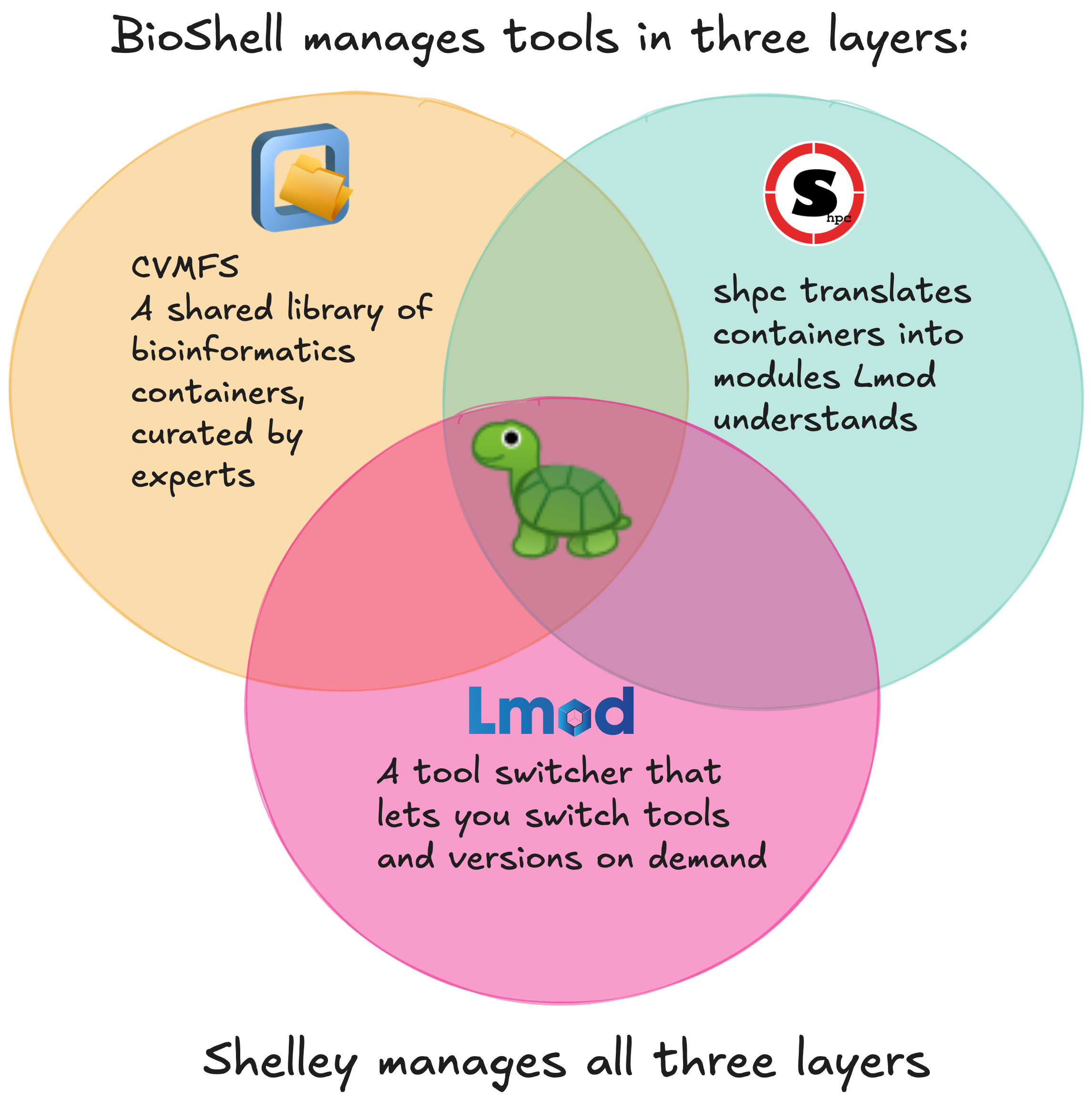
You don’t need to undersand any of this to use BioShell because Shelley, BioShell’s command-line assistant, drives all three for you. She:
- Searches the tool library
- Picks the right container version
- Creates any sHPC registry entry that is missing
- Installs the module: one command to find a tool, one to install it
Shelley basic usage 
Shelley runs from the command line or in an interactive mode. It can be used to manage your bioinformatics tool containers. We currently only support command-line tools and are working on extending this functionality out to R and Python packages.
Run Shelley with:
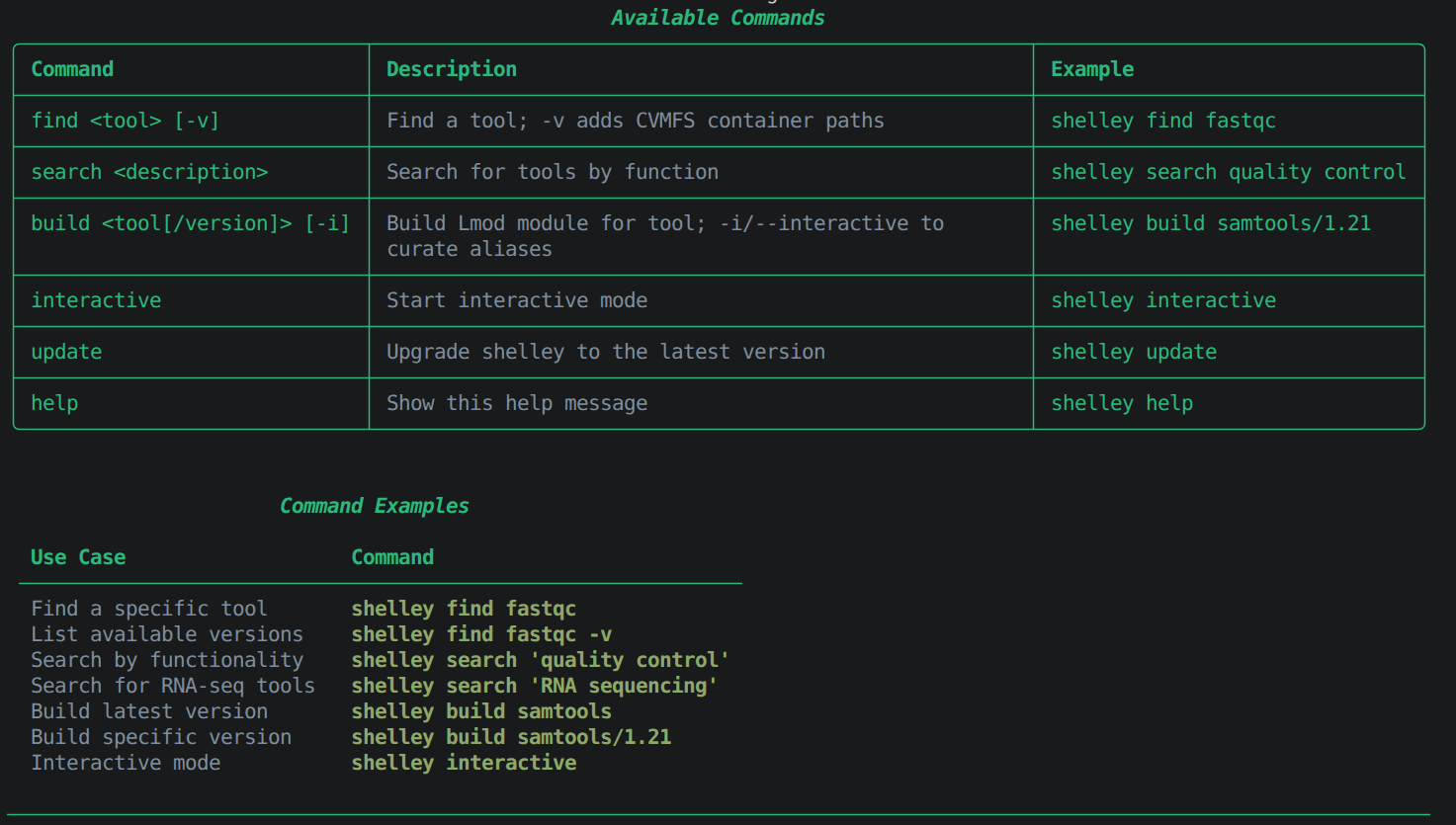
shelley help
Example output
From the command line:
shelley find <tool> # Look up a specific tool by name
shelley search "<function>" # Search by keyword or function
shelley build <tool> # Install the tool as a loadable module
In interactive mode:
shelley interactive # Launch Shelley in interactive mode
Interactive mode works the same way as the command line. The find, search, and build
behave identically, except you type just the command name and its arguments, without
prefixing every call with shelley.
Follow our Shelley tutorial to practice using Shelley to find, search, and build modules.
Reference genomes and indexes
Reference genome builds and pre-built indexes, managed and maintained by the Galaxy Project, sit in two directories:
ls /cvmfs/data.galaxyproject.org/byhand/ # by genome build, then index type
ls /cvmfs/data.galaxyproject.org/managed/ # by index type, then genome build
To use a reference file in your analysis, pass its absolute path directly to your tool or pipeline config. For example, the human CHM13 T2T v2.0 FASTA file is at:
/cvmfs/data.galaxyproject.org/byhand/CHM13_T2T_v2.0/seq/CHM13_T2T_v2.0.fa
The T2T genome directory also includes pre-built indexes for common aligners:
ls /cvmfs/data.galaxyproject.org/byhand/CHM13_T2T_v2.0/
# bowtie2_index/ bwa_mem_index/ bwameth_index/ hisat2_index/ len/ rnastar/ seq/
The reference datasets available through CVMFS are maintained by the Galaxy Project and may not be comprehensive.
Troubleshooting
CVMFS probe fails for a repository
The repository may be temporarily unavailable. Wait a moment and run cvmfs_config probe
again. Contact Australian BioCommons support if
the problem persists.
Module appears in module avail but will not load
Check that both shpc and singularity are loaded:
module list
sHPC requires Singularity to execute containers.
Shelley cannot find a tool
Try search with broader keywords, for example shelley search "alignment" instead of a
specific tool name. If the container exists in CernVM-FS but Shelley does not index it, install
the module manually with shpc install — see the
sHPC user guide.
Further reading
- How to use Shelley — snippets for various use cases
- Full CLI reference
- Design rationale
- BioContainers registry
- Galaxy Project CVMFS repositories
- CVMFS documentation
- sHPC user guide