Read alignment and quantification
Questions
- How to map sequencing reads to a reference genome?
- How to use the mapped alignments to quantify gene-counts
We’ve moved on to the second stage of the nf-core/rnaseq workflow: read alignment and quantification (red box below).
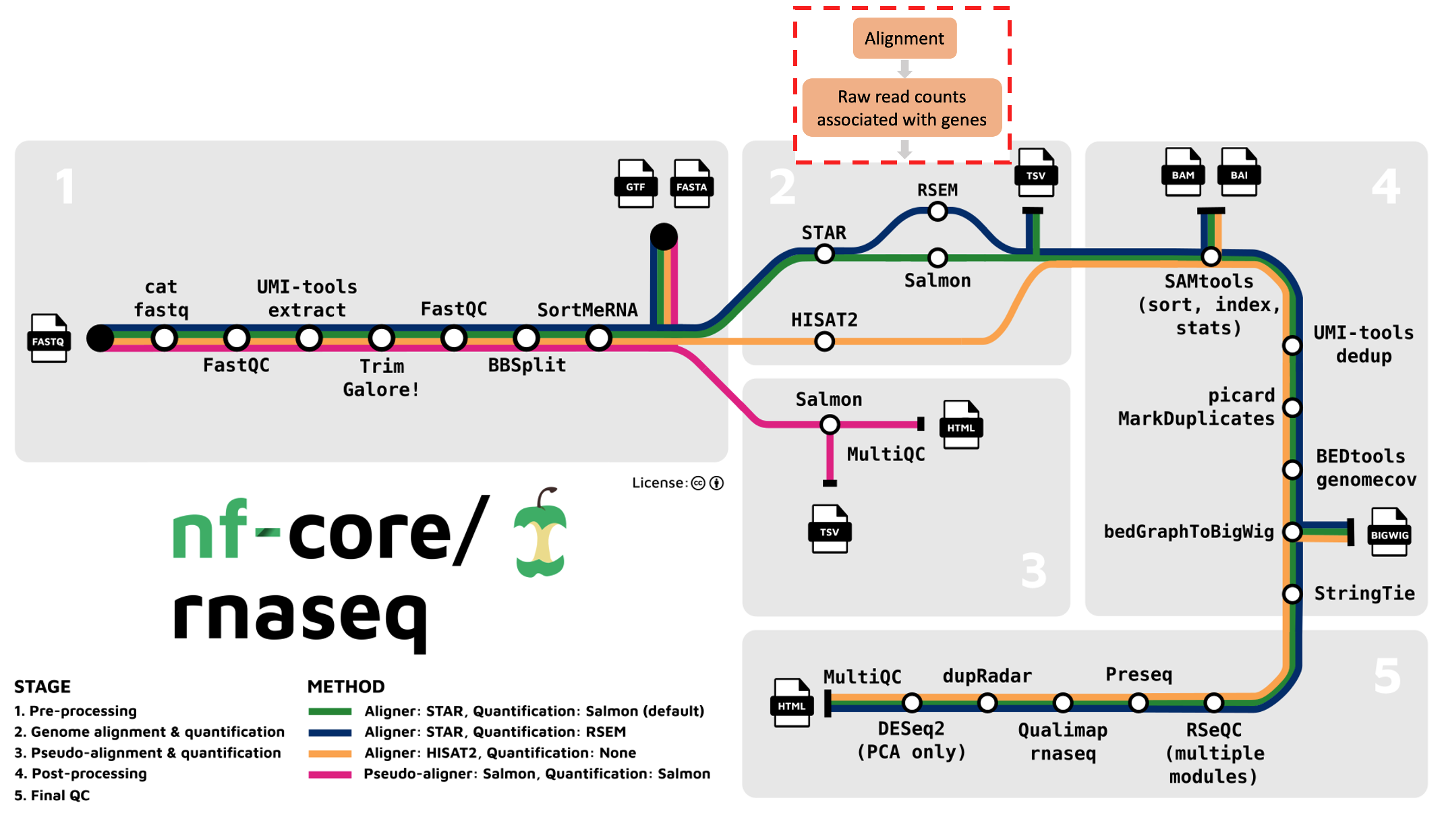
Alignment to reference genome
After read trimming, the nf-core/rnaseq pipeline maps reads to a reference genome specified by the user. Keep in mind that RNA-seq specific alignment tools are different to whole genome alignment tools. They are designed to be ‘splice aware’, meaning they are capable of differentiating intronic from exonic regions in the alignment process.
Bulk RNA-seq reads are derived from mature mRNA and contain only exons (and no introns). This means a sequence read can span 2 exons. Splice-aware aligners use a reference genome, not a transcriptome to perform read alignment, which contain intergenic, intronic and exonic sequences. This means that when they’re aligned to the reference genome, RNA-seq reads might span large introns. Splice aware aligners know not to align the RNA-seq reads to introns and can align a read across exons.
mRNA splicing is the process by which an mRNA transcript prepares to be translated into an amino acid sequence. It works by removing introns and splicing all exons back together to create a mature mRNA that is transported from the nucleus to the cytoplasm, ready to undergo translation.
The nf-core/rnaseq pipeline offers us various alignment and quantification routes:
- STAR – Salmon
- STAR – RSEM
- HISAT2 – no quantification
Aligned sequences for each sample are output in the bam file format.
The challenge activity for this session will be a group exercise. In breakout rooms, your facilitator will demonstrate how we can visualise alignments using a tool called IGV.
Challenge
This is a hard one! Thinking back to the case study (slide 13-14), can you work out how you would handle the scenario below?
Oh no! We have forgotten to label the conditional groupings of our samples and we don’t know which samples belong to the wildtype or knockout groups!
Can we use the alignment files to assign samples to their correct treatment group?
Solution
- From the case study, we know a loss of function mutation of Gtf2ird1 was generated by an insertion of a Myc transgene, resulting in a 40 kb deletion surrounding exon 1.
- Navigate to the gene in the mm10 assembly (chr5:134332897-134481480)
- Samples SRR3473984, SRR3473985, SRR3473984 contain reads covering exon 1. These are Wildtype samples.
- Samples SRR3473987, SRR3473988, SRR3473989 DO NOT contain reads covering exon 1. These are Knockout samples.
Read quantification
Following read alignment, we use the alignment files to calculate raw gene-count data for each sample. We can then use these count files (called gene-count matrix), to identify differentially expressed genes. By default, the nf-core/rnaseq pipeline runs Salmon for transcript quantification, following alignment by STAR.
Challenge
- Can you identify the per sample gene count files produced by STAR and Salmon?
- Can you identify the final gene count matrix created by STAR and Salmon?
Solution
You will see per sample salmon quant output files in sample directories, e.g results > star_salmon > SRR3473989 > quant.sf (transcript level) and quant.genes.sf (gene level). Columns of these files are described in the Salmon documentation. Note - raw counts is not a simple count of depth of reads, there is a lot more to consider and each tool does this slightly differently (e.g. how do you count reads spanning overlapping exons across two different genes?). See the Salmon documentation for how Salmon does it - this count is an estimation.
A count matrix file (tab separated values or TSV format) is also produced by the workflow. This looks very similar to what we will import in R tomorrow, it can be found here: results > star_salmon > salmon.merged.gene_counts.tsv. Most genes will have 0 counts because we subsetted the data.
As an example, you can find or grep the Dcc gene (ENSMUSG00000060534).This takes the number in “NumReads” for each sample and gene in the quant.sf file, and puts it into a count matrix file.
grep ENSMUSG00000060534 ~/base_directory/working_directory/results/star_salmon/salmon.merged.gene_counts.tsvProceed to the next lesson by clicking on What is nf-core/rnaseq doing? > Nextflow logs on the menu bar.
Key points
- Splice aware alignemnet tools like HISAT2 and STAR must be used when aligning RNA-seq reads to a reference genome.
- A variety of methods for read alignment and transcript quantification are available in the nf-core/rnaseq pipeline.
All materials copyright Sydney Informatics Hub, University of Sydney