Introduction to Nextflow
Questions
- Why use workflow management tools for bioinformatics?
- How does Nextflow simplify bioinformatics workflows?
- What are some of the benefits of using nf-core pipelines?
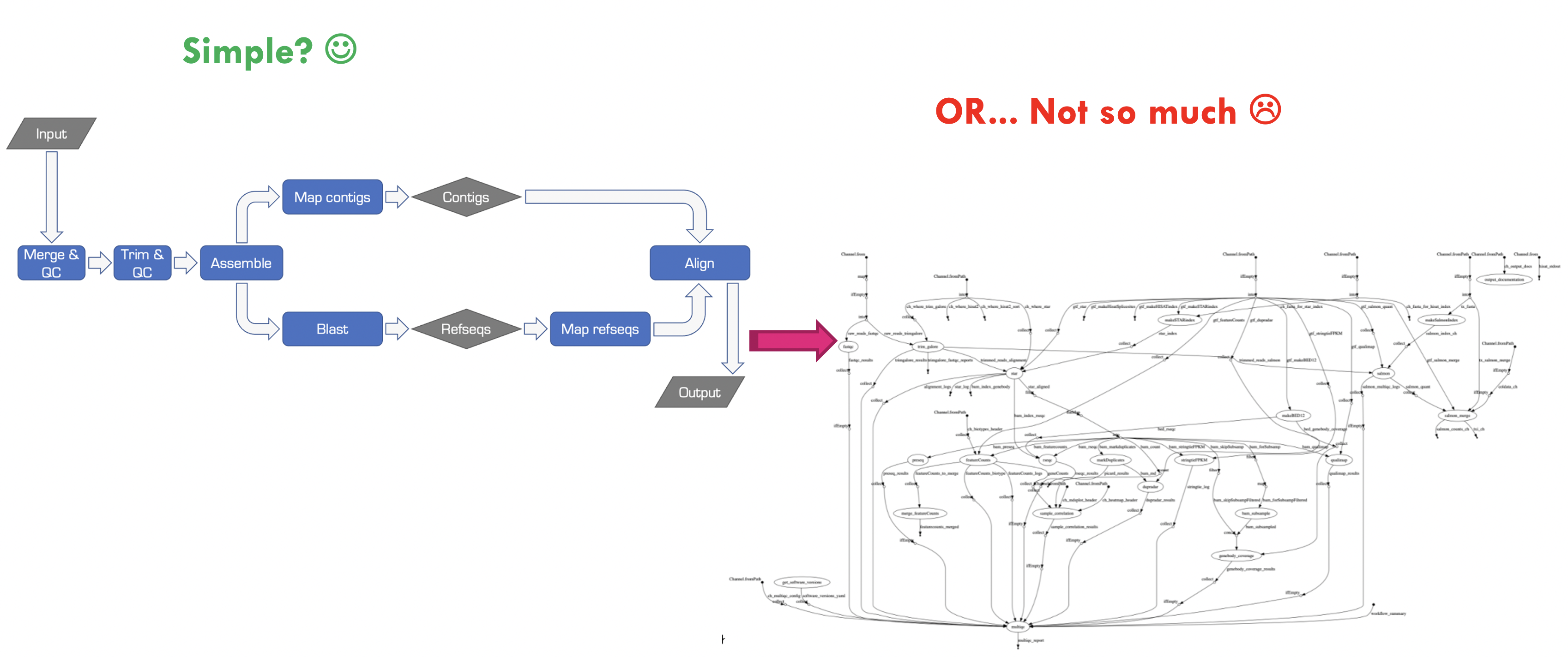
Bioinformatics workflows are like wet-lab protocols, they consist of many steps that need to be performed consistently across experiments. And just like wet-lab protocols, things can get very complicated very quickly when we’re working with different numbers of samples, different organisms, and collaborating with other researchers. Just take a look at the image below to see how messy things can get! When applied to a real dataset, the workflow on the left becomes a tangled mess when we break it down into all the individual tasks that are performed.
Bioinformatics workflows, especially those required to process next generation sequence (NGS) data (like RNA-seq), can be very repetitive. At the scale that biological data is growing, computational workflows are a great solution for performing these repetitive, tedious tasks.
What are workflow management systems?
Scientific workflow management systems can be used to automate analyses by stringing multiple processes together into a cohesive pipeline. Bioinformatics workflows often requires multiple processes, performed by multiple tools. As we apply NGS technologies to different organisms and research questions, we need these pipelines to be flexible and allow us to customise various parameters and integrate reference data dynamically.
Bioinformatics workflow managers are a great solution because they can:
- Simplify pipeline development
- Optimise resource usage
- Handle software installation
- Can run on different compute platforms
- Enable reproducibility and portability
Popular bioinformatics workflow managers include Snakemake, Nextflow, and Galaxy. These systems also provide a visual front-end, which allows the user to build and modify workflows without needing to have any prior programming expertise.
What is Nextflow?
Nextflow is one such workflow management tool. It is designed to address the challenges of building and running scalable, reproducible, and easy to use bioinformatics workflows.
Nextflow allows users to tie together various pieces of software and code written in different scripting languages and removes the need for software installation through the use of Conda, and container technologies like Singularity and Docker.
What is nf-core?
nf-core is a community curated collection of bioinformatics pipelines written in Nextflow (Ewels et al. 2020). The nf-core community is global, comprising bioinformaticians, computational biologists, software engineers, and biologists. The community works together to develop and maintain best practice bioinformatics pipelines and support others in running them. They’ve also developed a toolkit to assist in pipeline usage and development. Everyone is welcome to join the community!
Currently, 65 pipelines are available as a part of nf-core. Some of their most popular offerings include:
- nf-core/rnaseq for RNA-seq data obtained from organisms with a reference genome.
- nf-core/sarek for whole genome sequence data from organisms with a reference genome.
- nfcore-ampliseq for amplicon sequencing data including 16S, ITS, and 18S amplicons.
Proceed to the next lesson by clicking on Why use nf-core/rnaseq? > Introduction to nf-core/rnaseq on the menu bar.
Key points
- Nextflow is a workflow management tool designed to address the challenges of bioinformatics workflows.
- nf-core pipelines are community supported, open access, and easy to use.
All materials copyright Sydney Informatics Hub, University of Sydney