Raw read trimming
Questions
- Why trim RNA sequencing reads?
- How does Trim Galore work?
We’ve moved on to the second part of the first stage of the nf-core/rnaseq workflow: read trimming (red box below).
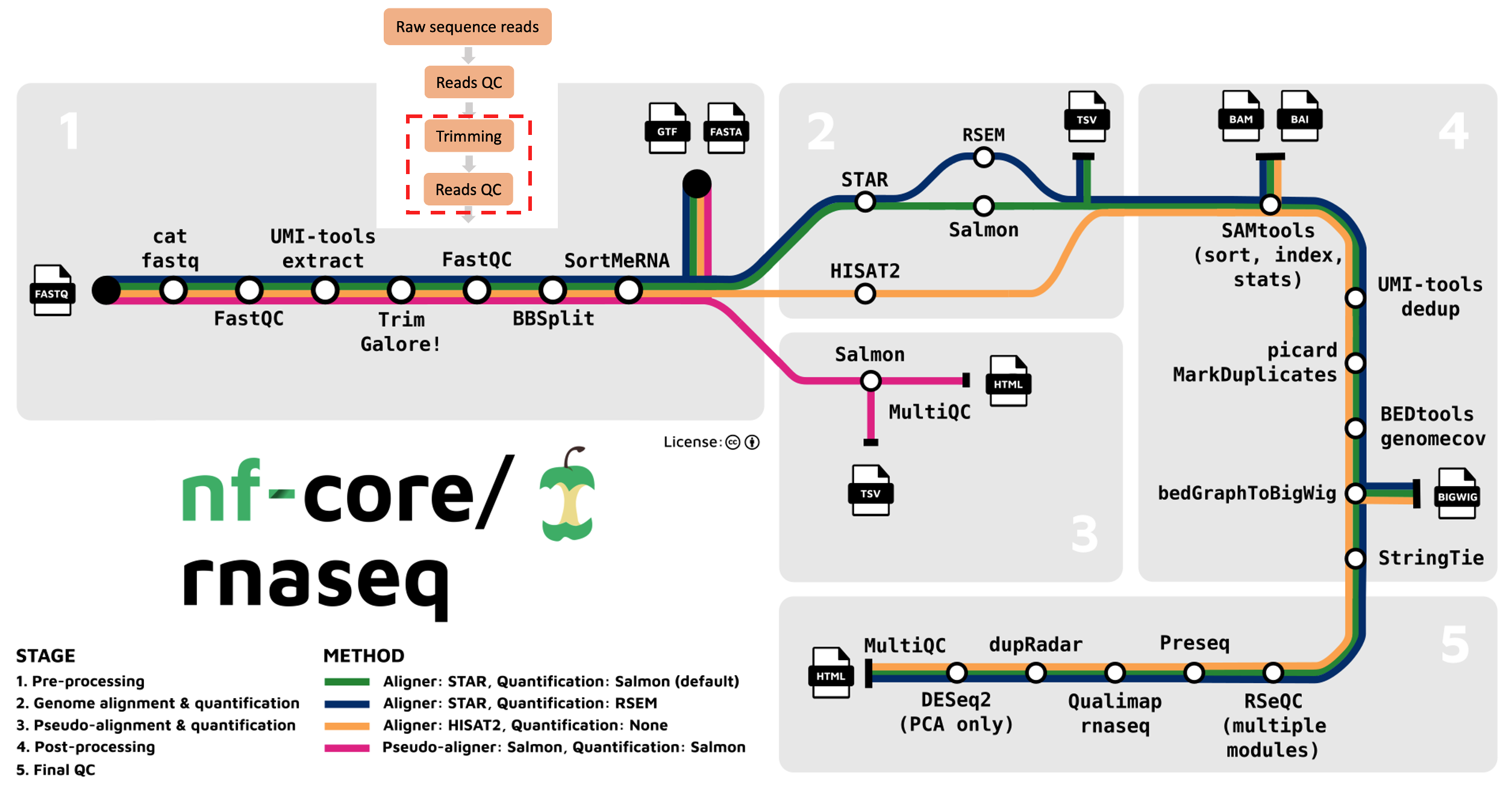
Why trim our sequence reads?
Trimming is sometimes performed to improve the quality of the raw data and potentially improve its mappability when it is being aligned to a reference genome. There are several ways to perform trimming:
- Removal of poor quality reads or bases (e.g. ends of reads)
- Removal of adapter sequences
- Removal of polyA tails
The nf-core/rnaseq pipeline uses Trim Galore for read quality trimming. It is able to perform quality-based removal of low-quality bases and adapter trimming. Given trimming can result in some reads being significantly shortened (sometimes to 0bp!), Trim Galore will filter reads that are too short to be used in downstream processes like read alignment.
Does trimming help?
Read trimming is not always a necessary step when processing next generation sequencing (NGS) data. These days NGS data is of a very high quality and the tools we use to perform processes like read mapping are capable of handling poor quality reads and adapter sequences.
While the trimming adapter sequences has been shown to increase the quality of RNA-seq data (Dozmorov et al., 2015), other studies have shown that trimming of poor quality reads can effect gene expression estimates (Williams at el., 2016).
When making the decision to trim your reads for differential expression RNA-seq studies, we suggest following the recommendations of the read alignment tool you’ll be using.
Open your Nimbus terminal again to do the challenge exercise below:
Challenge
Navigate to the results directory ~/base_directory/working_directory/results and answer the following questions:
- Which tool does nfcore-rnaseq use for read-trimming?
- Which tool did you use to generate quality reports before and after trimming?
- What effect did trimming have on SRR3473989.fastq?
Solution
Trim-galore is used for trimming.
FastQC generates .html reports.
Open the trimgalore report with:
cat trimgalore/SRR3473984.fastq.gz_trimming_report.txt. Also open the html files generated by fastqc inside the foldertrimgaloreon your local computer.- Total sequences has gone down
- Read length is now 21 - 101
- Per base sequence quality now mostly in the green
Proceed to the next lesson by clicking on What is nf-core/rnaseq doing? > Read alignment and quantification on the menu bar.
Key points
- Tools such as Trim Galore can be used for sequence read trimming.
- We should evaluate our data before deciding to trim reads.
- Confirm what kind of read trimming is required (if any) by the alignment tool you’re using.
All materials copyright Sydney Informatics Hub, University of Sydney