Example parallel job
Overview
Teaching: 15 min
Exercises: 15 minQuestions
Run multi-node parallel jobs on Gadi
Objectives
Use OpenMPI and nci-parallel to run parallel jobs
Example parallel jobs
Below we have two examples: one for bioinformatics and one for modelling/simulation. During live delivery of this workshop, we will have separate breakout rooms for each example.
Bioinformatics example
Following on from our DNA example, we have two samples that we will map to the reference genome. For training purposes, the data has been downsized and the reference ‘genome’ restricted to one chromosome.
Each sample has paired-end sequence data from two lanes of a sequencing flowcell, yielding four files per sample. There are around 10 million reads per file. Thinking about the way mapping works, we can see that this is perfectly suited to parallel execution, as each pair of reads are mapped independently of all other pairs in the dataset. We could theoretically execute this mapping in 40 million parallel tasks - however the time to execute the mapping command and load the reference index would exceed the time to map a single read pair, so this task size is too small. Through prior testing at SIH, we split fastq data into 500,000 reads per pair and this gives a mapping time of ~ 2 - 4 minutes, depending on reference genome and data quality.
So for this mapping example, to produce a BAM file per sample, we need to perform 3 jobs:
- Physically split the input data
- Perform the mapping in parallel
- Merge the parallel mapping output to one BAM per sample
For some bioinformatics tasks (eg GATK variant calling or base quality score recalibration) we do not need to physically split data, we can use other methods to define genomic intervals over which to operate. See our SIH bioinformatics repo for examples of this.
Sample data
You should already have the Sample_data_bio and Sample_data_bio_fastq datasets downloaded ready for this section. If not, please revist the download steps here.
You probably unpacked Sample_data_bio during the benchmarking section, please go ahead and unpack Sample_data_bio_fastq if you haven’t already done so:
tar -zvxf Sample_data_bio_fastq.tar.gz
Move the contents of Sample_data_bio_fastq to Sample_data_bio/Fastq then change directory:
mv Sample_data_bio_fastq/* Sample_data_bio/Fastq
cd Sample_data_bio
Fastq data for two samples is now in Fastq. This is important to ensure compatibility of filepaths in the sample scripts. Both samples were obtained from NCBI SRA (BD401; BD514). The required reference files are in Reference and scripts are in Scripts.
Required software
The example workflow will use some global apps (SAMtools, BWA, OpenMPI and nci-parallel) and two local apps (fastp and sambamba). You will need to have your own local installations of fastp and sambamba. To expedite this, an ‘apps’ directory with compiled versions of the software along with the required module files has been included in the Sample_data_bio package.
Please move the apps directory to /scratch/<project>:
mv Sample_data_bio/apps /scratch/<project>
Note that all of the steps described in section 6 Software have been done except for the last which requires you to add the following line to your .bashrc and .bash_profile files:
export MODULEPATH=${MODULEPATH}:/scratch/<project>/apps/Modules/modulefiles
You will also need to update the project code within the two .base files to your specific project code, eg
sed -i 's|/scratch/er01|/scratch/<project>|g' /scratch/<project>/Modules/modulefiles/fastp/.base
sed -i 's|/scratch/er01|/scratch/<project>|g' /scratch/<project>/Modules/modulefiles/sambamba/.base
Once you have moved apps to the appropriate location and edited these four files, from a new session (or run ‘source’ over the profile files), test the software is now available to use with module commands:
module load fastp/0.20.0
module load sambamba/0.7.1
fastp --version
sambamba --version
![]() Please ask for assistance if these commands failed
Please ask for assistance if these commands failed
Parallel method explained
Open Scripts/split_fastq_run_parallel.pbs and update the -P and -lstorage directives to your project code. You do not need to make any other changes to this script.
This is the standard “run” script that we use at SIH to launch parallel tasks on Gadi. Distribution of the tasks across nodes is handled by OpenMPI and the NCI custom wrapper script nci-parallel.
For each line of $INPUTS, $SCRIPTS is launched, taking in the parameters on that line as arguments to $SCRIPT. The number of tasks launced at once is dictated by the values at $NCPUS and at #PBS -l ncpus=<total_NCPUS>, ie if you request 96 total CPUs for the job and 2 CPUs per parallel task, 48 tasks will be executed simultaneously.
Tasks are executed in the order in which they appear in $INPUTS. Once a task is complete, those now-free CPU will be assigned to the next task in the list, until all tasks are complete.
The $SCRIPT file must be executable or the job will fail and exit. All non-existing directories required by $SCRIPT should be made in the run script. Modules required by $SCRIPT should also be loaded here to reduce overhead.
The $INPUTS file can contain any number of or type of parameter that your analysis requires. $SCRIPT must parse in these parameters, splitting out by delimiter in the event that there are multiple parameters. For our first job ‘split fastq’, there is only one parameter (the prefix of the fastq pair to split) so the bash positional variable $1 can be used to read the fastq prefix from $INPUTS into $SCRIPT.
![]() Take a moment to view the three files associated with this first job, and please feel free to ask any questions:
Take a moment to view the three files associated with this first job, and please feel free to ask any questions:
Scripts/split_fastq.shScripts/split_fastq_run_parallel.pbsInputs/split_fastq.inputs
Job 1: Split fastq
For this job, we have four inputs, and require 4 CPU per parallel task. This means we need to request 16 CPUs for the job, and 64 GB RAM (16 X 4).
Note that requesting less than one whole node worth of CPUs using this parallel method is OK, however if you require more than one node worth of CPUs, you must request a whole node. Ie if you require 80 CPUs, you must request 96 CPUs. Understand that this will decrease your CPU efficiency due to the 16 idle nodes. In the next job, we will look at how to help minimise idle CPUs in multi-node jobs.
No changes to the scripts are required, so please go ahead and submit the job (from the Sample_data_bio working directory):
qsub Scripts/split_fastq_run_parallel.pbs
![]() Try out the commands
Try out the commands nqstat_anu and qps <jobID> (quickly, job will complete in ~ 1 minute).
Once complete, check your Logs:
grep Exit Logs/split_fastq.o
grep "exited with status 0" Logs/split_fastq.e | wc -l
Note: as at 22/01/21, there is a small issue with these parallel jobs printing two ‘UCX WARN’ error messages per job to the .o log. NCI is aware of this and these can safely be ignored.
Check your output directory, it should now contain a number of small fastq files of around 40 MB each:
ls -1 Fastq_split/ | wc -l
The number of split outputs should equal double the length of the inputs list.
By splitting the input, we have increased the level of parallelism possible for this mapping from 4 to ~ 88.
![]() For a large dataset, you can imagine that this method which doubles your disk and iNode usage can quickly push your project limits, so active management is required! (Recall the
For a large dataset, you can imagine that this method which doubles your disk and iNode usage can quickly push your project limits, so active management is required! (Recall the lquota command).
Job 2: Align
Next we will map/align the split fastq to the small reference. Alignment requires more parameters than data splitting, as there is a lot of metadata required for the output BAM file.
View the sample configuration file test_samples.config. This contains the metadata we want to include in the output. The script Scripts/align_make_input.sh reads this file and the output of the ‘split fastq’ job to create the inputs file.
Make the inputs file, parsing the prefix of your config file as argument:
bash Scripts/align_make_input.sh test_samples
![]() View the inputs file format, and lines 37 - 46 of the script file:
View the inputs file format, and lines 37 - 46 of the script file:
head -1 Inputs/align.inputs
cat -n Scripts/align.sh
This is one example of how you can make large and multi-parameter inputs files, but ultimately how you make your inputs will depend on your specific analysis and preferred methods.
Using the parallel resource calculator
We have ~ 88 tasks to run. Revisit your earlier ‘align’ benchmarking results: we know we want to use 4 CPUs per task, and expect a walltime of ~ 2.5 minutes.
To select our resources for this job, download the parallel resoure calculator to your computer and open in Microsoft Excel.
Edit the values in cells A3 to A7 to match your analysis. Try different node numbers at cell A4 and view the impact on walltime at cell A17.
Observe the value of A14 ‘chunks of tasks’ as you change number of nodes. Anything less than 1 will result in idle CPU, and reduce your CPU efficiency. For jobs where the task time is expected to be fairly constant among tasks (like this one, as our fastq files are all ~ 500,000 reads) a value close to but just under an integer is desired and most likely to maximise CPU efficiency. If however your task times are expected to be variable, and you have ordered your inputs by largest to smallest expeted walltime, this is not the case - a bit of maths could help you work out the ideal “chunks of tasks” value but that is beyond the scope of this workshop!
![]() How many nodes would you request for this job?
How many nodes would you request for this job?
Back to job 2…
Before submitting, run qstat -q. How does the normal queue look? You might consider changing the queue to ‘express’, ‘normalbw’ or ‘expressbw’ - completely up to you, just don’t forget to update the queue name in the calculator at cell A5!
Note that the ‘run_parallel’ PBS script auto-detects the number of CPUs per node based on the queue you have submitted to (under ‘Do not edit below this line’). This makes it easier for you to quickly modify your job configurations for different queues. You just need to ensure that the CPUs per task you assign via the ‘NCPUS’ variable is divisible by the number of CPUs per node in your chosen queue (28 for Broadwell queues, 48 for others).
Update your PBS -l directives for waltime, total cores and total mem based on the number of nodes and queue you have selected. Then submit the job (from your Sample_data_bio directory):
qsub Scripts/align_run_parallel.pbs
Check/monitor:
qstat -u <userID>
nqstat_anu
qps <jobID> | more
Once complete, check your Logs and output:
grep Exit Logs/align.o
grep "exited with status 0" Logs/align.e | wc -l
ls -1 Align_split/ | wc -l
The number of tasks which exited with status 0 should equal the number of inputs should equal the number of BAM files in ./Align_split.
Additional checks include looking for reported errors in the ‘Error_capture/Align_split directory, and sorting the BAM files by size to look for empty/too small BAM files.
Report job usage metrics:
cd Logs
perl ../Scripts/gadi_usage_report.pl align
| #JobName | CPUs_requested | CPUs_used | Mem_requested | Mem_used | CPUtime | CPUtime_mins | Walltime_req | Walltime_used | Walltime_mins | JobFS_req | JobFS_used | Efficiency | Service_units | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| align | 384 | 384 | 1.48TB | 95.28GB | 8:59:56 | 539.93 | 0:04:30 | 0:02:03 | 2.05 | 800.0MB | 8.17MB | 0.69 | 26.24 |
![]() How does your CPU efficiency compare to that from the benchmarking run at NCPUS=4?
How does your CPU efficiency compare to that from the benchmarking run at NCPUS=4?
Hopefully it is not too much lower. If it is, this could mean we picked a non-representative sample for benchmarking - ideally, you should benchmark on multiple samples to guard against this. Also to consider is the overhead of each task being executed, and all the processes per task that are launched - these may contribute only milliseconds each, but as the level of parallelism increases, this adds up and does yield a reduction in CPU efficiency.
Special note for parallel GATK
On Gadi we see a particular decline in CPU efficiency for GATK parallel jobs. Sadly this only occured after a recent Gadi system upgrade and we have yet to design a better solution for parallel GATK. It is to do with how GATK essentially thrashes the Lustre filesystem. We intend to explore the use of jobfs but for mammalian genomics, the size of the data to be copied back and forth between jobfs means this is unlikely to improve performance. Currently, we are managing this by capping the number of GATK tasks running at once to 200. If you follow our Fastq-to-BAM, Bootstrapping-for-BQSR, Germline-ShortV or Somatic-ShortV optimised Gadi pipelines, please note that these were developed prior to onset of the issue and as such your walltime and CPU efficiency is likely to be worse than the examples noted.
Job 3: Merge the alignment output
We now need to take the ~ 88 BAM files and merge these into two sample-level BAMs. Like splitting the fastq, this may be an extra step compared to just processing the whole original fastq in one alignment job per sample - however the sum of time for these three jobs is less than the walltime for one non-parallel alignment job typically by ~ 6x. For our dataset however (like many whole genome sequencing projects) a merge step would have been required anyway, as the data must be aligned at a per-lane-per-sample level, and our two samples each had two lanes.
Make the inputs file:
bash Scripts/merge_align_make_input.sh test_samples
Then submit the job:
qsub Scripts/merge_align_run_parallel.pbs
Check/monitor:
qstat -u <userID>
nqstat_anu
qps <jobID>
Once complete, check your Logs and output:
grep Exit Logs/merge-align.o
grep "exited with status 0" Logs/merge-align.e | wc -l
ls -lh Align_merged
Additional checks include looking for reported errors in the Error_capture/Merge_align directory, and checking for empty/too small BAM files.
Report job usage metrics:
cd Logs
perl ../Scripts/gadi_usage_report.pl merge_align
Parallel example wrap-up
- Gadi scheduler favours ‘short and wide’ jobs, so where possible, split your data (either physically or by defining intervals/ranges) to enable parallelism
- Key bioinformatics tasks like mapping and short variant calling are ideally suited to this
- Use OpenMPI and nci-parallel method to distribute tasks listed in
$INPUTSto job commands in$SCRIPTSby using a ‘parallel run’ script - Use the parallel resoure calculator to help size your job
- Perform benchmarking prior to setting up a parallel job
- Use representative samples/inputs
- Ideally, benchmark on multiple samples/inputs to capture variation
- Assess how well your job scales by looking at the CPU efficiency, that can be reported using the gadi usage report script
- Before executing parallel jobs, it is beneficial to run some saclability tests after your benchmarking, eg if your parallel job will have 100,000 tasks, run subset jobs of say 100 and 1,000 tasks first: does the CPU efficiency remain stable as nodes and simultaneous tasks increases? If not, consider the impact this will have on your walltime (allowing a 1.5 X buffer won’t be enough!) and explore ways in which CPU efficiency might be improved for at-sacle runs.
End of session
![]() Does anyone have any questions about this example parallel job, or other aspects they would like to discuss in more detail?
Does anyone have any questions about this example parallel job, or other aspects they would like to discuss in more detail?
![]() Thank you all for your attendance and participation! We hope you have found this session valuable, and welcome any feedback via our survey.
Thank you all for your attendance and participation! We hope you have found this session valuable, and welcome any feedback via our survey.
Happy high performance computing! ![]()
Simulation example
Python multiprocessing
What is MPI
MPI is a standardized and portable message-passing system designed to function on a wide variety of parallel computers. The standard defines the syntax and semantics of a core of library routines useful to a wide range of users writing portable message-passing programs in C, C++, and Fortran. There are several well-tested and efficient implementations of MPI, many of which are open-source or in the public domain.
Most software that use MPI in the modelling and simulation space is enabled via running the application with mpirun and specifying the number of processes. For instance, fluid dynamic applications like openfoam allow running the solver with mpi in mind. These however are quite specific to the research domain, and take time to set up configuration files and conduct pre-processing. Instead, we will look at a basic python program, and how to use a package called multiprocessing to run generic scripts with MPI in mind.
2 packages that enable multiprocessing in python are mpi4py and the multiprocessing library. The former is not installed on gadi, so we will focus on using the latter.
With multiprocessing, Python creates new processes. A process here can be thought of as almost a completely different program, though technically they are usually defined as a collection of resources where the resources include memory, file handles and things like that.
One way to think about it is that each process runs in its own Python interpreter, and multiprocessing farms out parts of your program to run on each process.
Some terminology - Processes, threads and shared memory
A process is a collection of resources including program files and memory, that operates as an independent entity. Since each process has a seperate memory space, it can operate independently from other processes. It cannot easily access shared data in other processes.
A thread is the unit of execution within a process. A process can have anywhere from just one thread to many threads. Threads are considered lightweight because they use far less resources than processes. Threads also share the same memory space so are not independent.
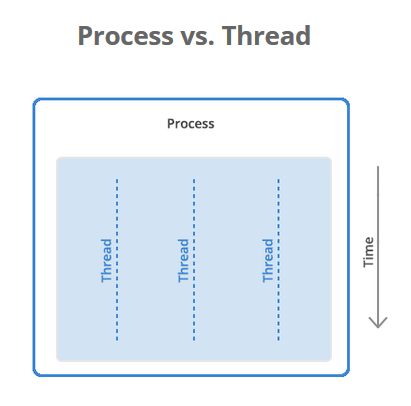
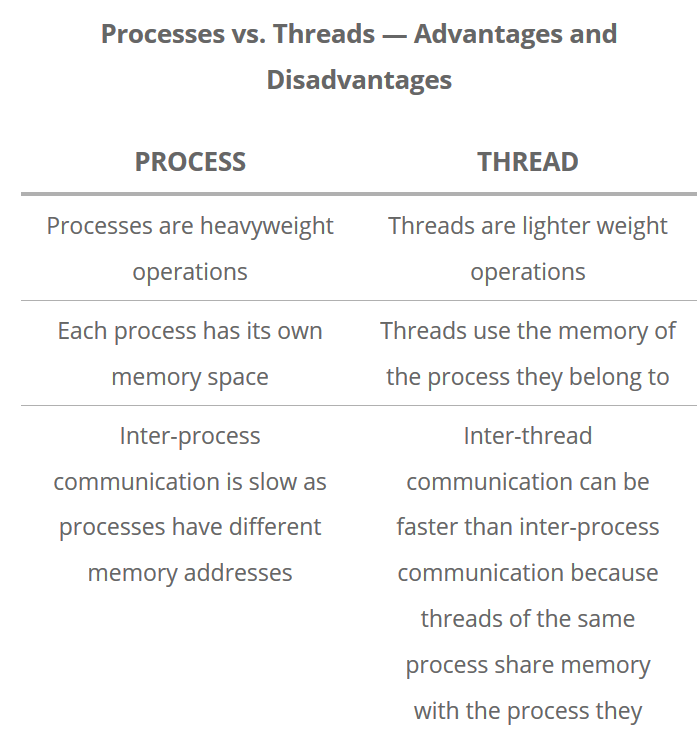
Back to python, the multiprocessing library was designed to break down the Global Interpreter Lock (GIL) that limits one thread to control the Python interpreter.
In Python, the things that are occurring simultaneously are called by different names (thread, task, process). While they all fall under the definition of concurrency (multiple things happening anaologous to different trains of thought), only multiprocessing actually runs these trains of thought at literally the same time. Low level primitives exist that organise sharing info between processes (pipes and queues), but we will stick to high level stuff.
Experience with MPI and nci-parallel
Scripts that will be covered can be downloaded in the below link for future reference. Other supportive documentation links on this subject are also given.
Extra NCI documentation on OpenMPI
Extra NCI documentation on nci-parallel
The file whatispi_mpi.pbs holds a few variations on running a python script in different ways. The underlying python file whatispi.py runs a simulation on estimating the number pi given a set of numbers - The higher the simulation number the closer the result should be to the true value of pi.
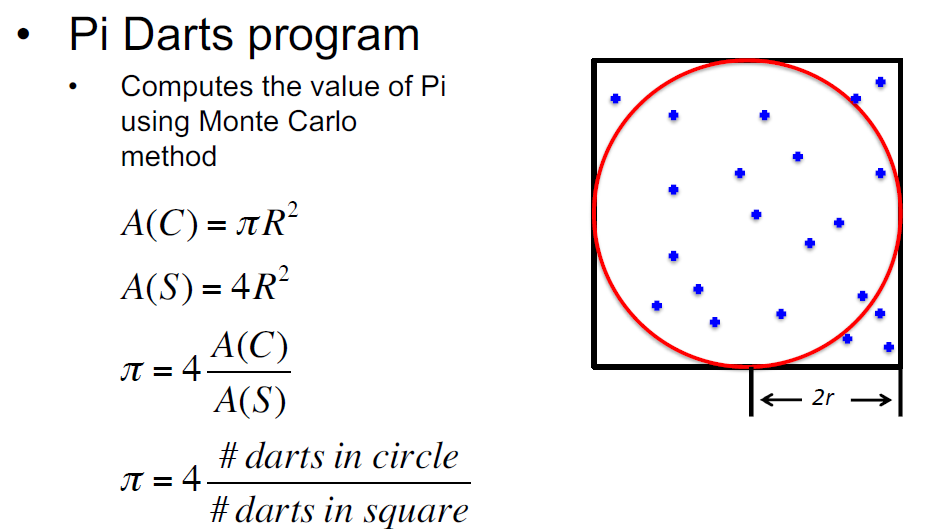
Within the whatispi.py file, some basic concepts that use the multiprocessing library are:
- the
Pool(processes)object creates a pool of processes.processesis the number of worker processes to use (i.e Python interpreters). IfprocessesisNonethen the number returned byos.cpu_count()is used. - The
map(function,list)attribute of this object uses the pool to map a defined function to a list/iterator object
Open-MPI usage
There are a few ways to use MPI with your program on Gadi. The simpliest way is a generic mpirun after your PBS directives such as:
module load openmpi/4.0.3
mpirun ./a.out > output
Or Specify the number of processes / copies of the program
mpirun -np N <program>
If no value is provided for the number of copies to execute, Open MPI will automatically execute a copy of the program on each process slot. Manual page for mpirun options
More specific implementations are done via passing extra arguments with the -map-by flag. Some examples of this are given below, however summarising extra terminology specific to this command is useful for understanding.
MPI argument definitions
–map-by <obj>:PE=n - This maps specified physical objects / resources. Supported options include slot, hwthread, core, L1cache, L2cache, L3cache, socket, numa, board, node, sequential, distance, and ppr. Any object can include modifiers by adding a : and any combination of PE=n
ppr - Comma-separated list of number of processes on a given resource type
ppr:N:resource - where N is a number and resource related to ype of physical compute. Rsource defaults to socket. Some options are socket, node, numa, etc
N - number
PE - Bind each process to the specified number of cpus
A variety is given below which is within the pbs file. Various implementations are demonstrated now from the whatispi_mpi.pbs, and a summary of them is below. Confirm your understanding of different specifications with the pbs output files.
Method 1:Basic usage
mpirun python whatispi.py 1 1000 5000 10000
Method 2: Specifying number of processes
mpirun -np 2 python whatispi.py 4 1000 5000 10000
Method3: Passing further process paralisation to python
mpirun -np 2 python whatispi.py 4 1000 5000 10000
Method4: Mixed Mode - MP between nodes and OMP threads within.
n=2
export OMP_NUM_THREADS=$n
mpirun -map-by ppr:$((8/$n)):numa:PE=$n python whatispi.py 1 1000 15000
Method5:More practice on specifying resources
mpirun -map-by ppr:2:node python whatispi.py 1 100000 2000000
nci-parallel
The module nci-parallel provides a MPI-based taskfarming application to run embarrassingly parallel tasks in a single big PBS job. It maintains its own task queue, every time there are subtasks finished in the PBS job, the next tasks in the list will be dispatched to the available cores.
Within the downloaded scripts, the cmds.txt file holds a series of mpi commands that are executed in parallel, and the parallel.pbs script uses the mpirun command with an extra nci-parallel flag with it start the process. To use nci-parallel on gadi, a general description after PBS declarations is below, extracted from nci documentation.
module load nci-parallel/1.0.0
export ncores_per_task=4
export ncores_per_numanode=12
mpirun -np $((PBS_NCPUS/ncores_per_task)) --map-by ppr:$((ncores_per_numanode/ncores_per_task)):NUMA:PE=${ncores_per_task} nci-parallel
--input-file cmds.txt --timeout 4000
nci-parallel - flag within the mpirun command that engages nci-parallel
timeout - controls the maximum execution time for every task wrapped in the job
input - specifies the list of commands to run
End of session
![]() Does anyone have any questions about this example parallel job, or other aspects they would like to discuss in more detail?
Does anyone have any questions about this example parallel job, or other aspects they would like to discuss in more detail?
![]() Thank you all for your attendance and participation! We hope you have found this session valuable, and welcome any feedback via our survey.
Thank you all for your attendance and participation! We hope you have found this session valuable, and welcome any feedback via our survey.
Happy high performance computing! ![]()
Key Points
As Gadi does not allow job arrays, we recommend OpenMPI and nci-parallel to distribute parallel tasks over CPUs/nodes
Included examples can hopefully be adapted for your own work