Recap of day 1
Questions
- What stages of the RNA-seq workflow does the nf-core/rnaseq pipeline perform?
- What were results and files were created by nf-core/rnaseq?
On day 1 of this workshop series, we used the nf-core/rnaseq pipeline to process raw sequence reads to generate a gene count matrix. We learned what steps are involved in a typical differential expression RNA-seq workflow. While we ran the default nf-core/rnaseq pipeline, we also discussed how to customise the pipeline’s run command depending on our own needs. Finally, we learned how to use quality control reports and alignment files generated by nf-core/rnaseq to evaluate and explore our data.
Given the computational resources and time required to run this pipeline on the mouse genome, we worked only with a subset of the real data. Today, we’ll be working with a whole genome count matrix, that has already been prepared. Today’s sessions will be a lot more hands on as we explore our gene count data, perform differential expression analysis, and enrichment analysis.
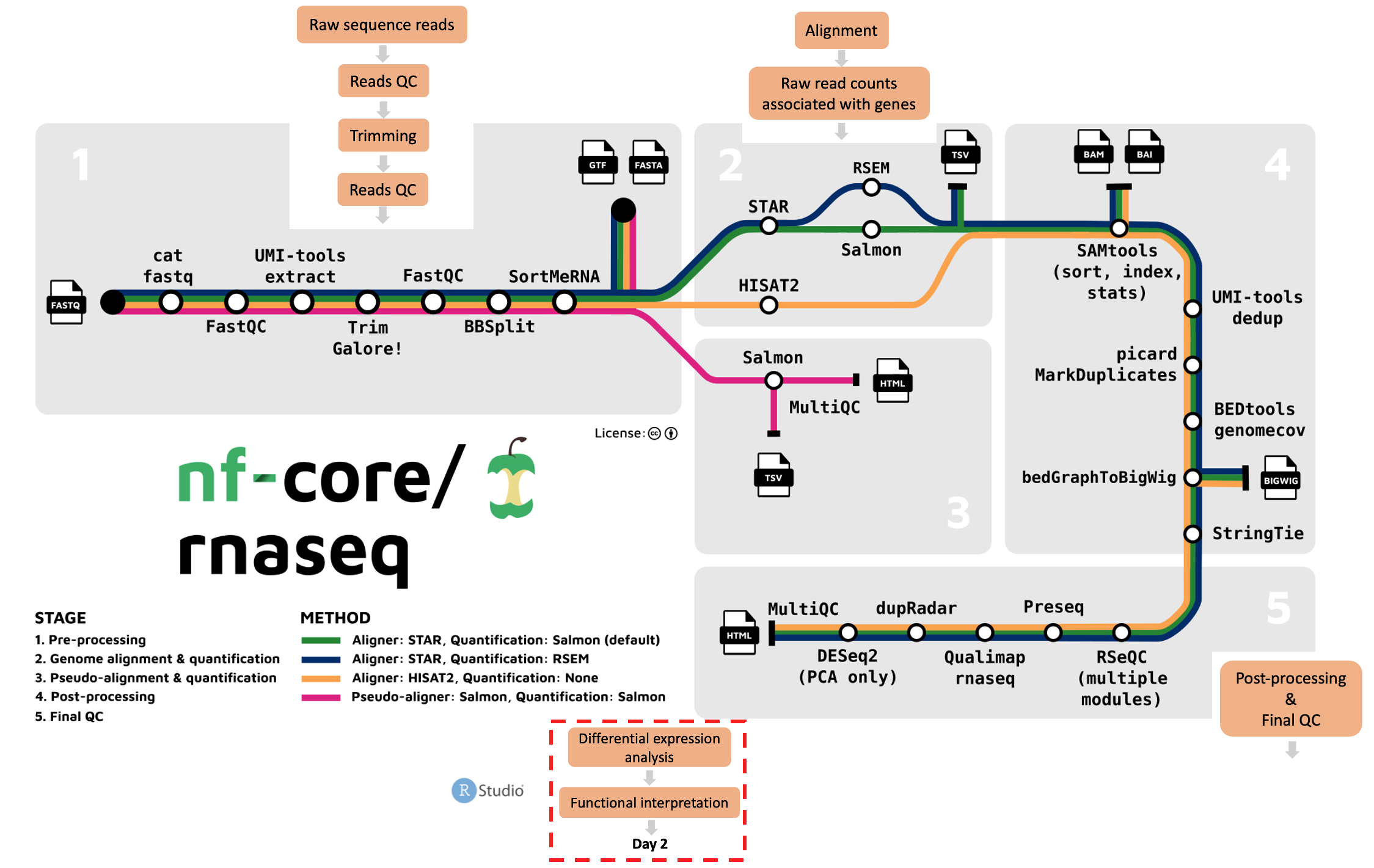
What’s the plan for day 2?
We will perform differential expression and enrichment analysis in R/Rstudio, launched from Nimbus. We will use a Singularity container that contains R/Rstudio with pre-installed libraries. We like using containers because they can be shared and run elsewhere, making your analysis portable and reproducible.
We’ll be using a prepared R markdown file to work through today’s activities. You can find at:
/home/training/base_directory/working_directory/rnaseq_DE_analysis_Day2.RmdAll materials copyright Sydney Informatics Hub, University of Sydney