Tips and tricks
VS Code: installing the code command
VS Code has a handy command-line tool called code that you can run to open any file or folder within VS Code. It comes pre-installed on our VMs, but if you are working on a machine without it, you can install it as follows:
- Open the command palette (
Ctrl + Shift + P/Cmd + Shift + P) and search for "Shell Command: Install 'code' command in PATH". When you find the command, click on it to begin the installation. - Close any active terminals within VS Code and start a new one for the changes to take effect.
Installing Nextflow
Nextflow is distributed as a self-installing package and can be installed using a few easy steps:
- Download the executable package using either
wget -qO- https://get.nextflow.io | bashorcurl -s https://get.nextflow.io | bash - Make the binary executable on your system by running
chmod +x nextflow. - Move the
nextflowfile to a directory accessible by your$PATHvariable, e.g.~/.local/bin/
Cleaning up the work directory
Your work directory can get very big very quickly (especially if you are using full sized datasets). It is good practise to clean your work directory regularly. Rather than removing the work folder with all of it's contents, the Nextflow clean function allows you to selectively remove data associated with specific runs.

Note that to prevent unintended deletion of important data, nextflow clean requires you to supply either the -dry-run or -force flag. By running nextflow clean -dry-run, you will see a list of files that would be removed if you were to instead provide the -force flag.
Additionally, the -after, -before, and -but options are all very useful to select specific runs to clean.
Listing and dropping cached pipelines
When using nextflow pull and nextflow run to automatically pull pipelines from their GitHub repositories, they will be cached locally within your $HOME/.nextflow/assets directory. Over time, these will build up and you might want to clean this directory up. Nextflow has functionality to help you to view and remove these cached pipelines.
The nextflow list command prints the projects stored in your cache folder. If you want to remove a specific pipeline from your cache you can remove it using the Nextflow drop command:
nf-core tools
Installing nf-core tools
nf-core tools is written in Python and is available from the Python Package Index (PyPI):
Alternatively, nf-core tools can be installed from Bioconda:
nf-core pipelines list
The nf-core pipelines list command can be used to print a list of remote nf-core pipelines along with your local pipeline information.
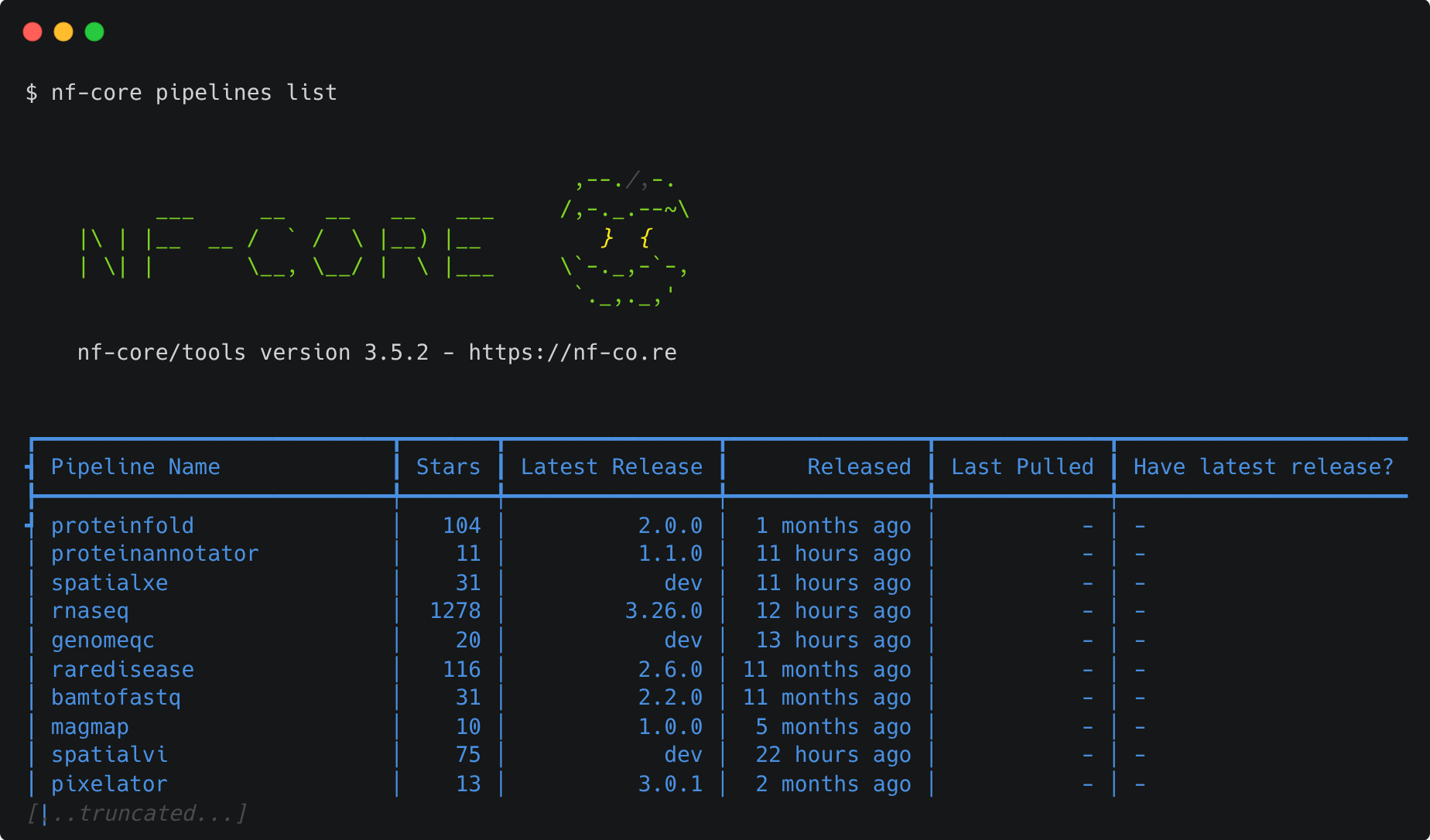
The output shows the latest pipeline version number and when it was released. You will also be shown if and when a pipeline was pulled locally and whether you have the latest version.
Keywords can also be supplied to help filter the pipelines based on matches in titles, descriptions, or topics:
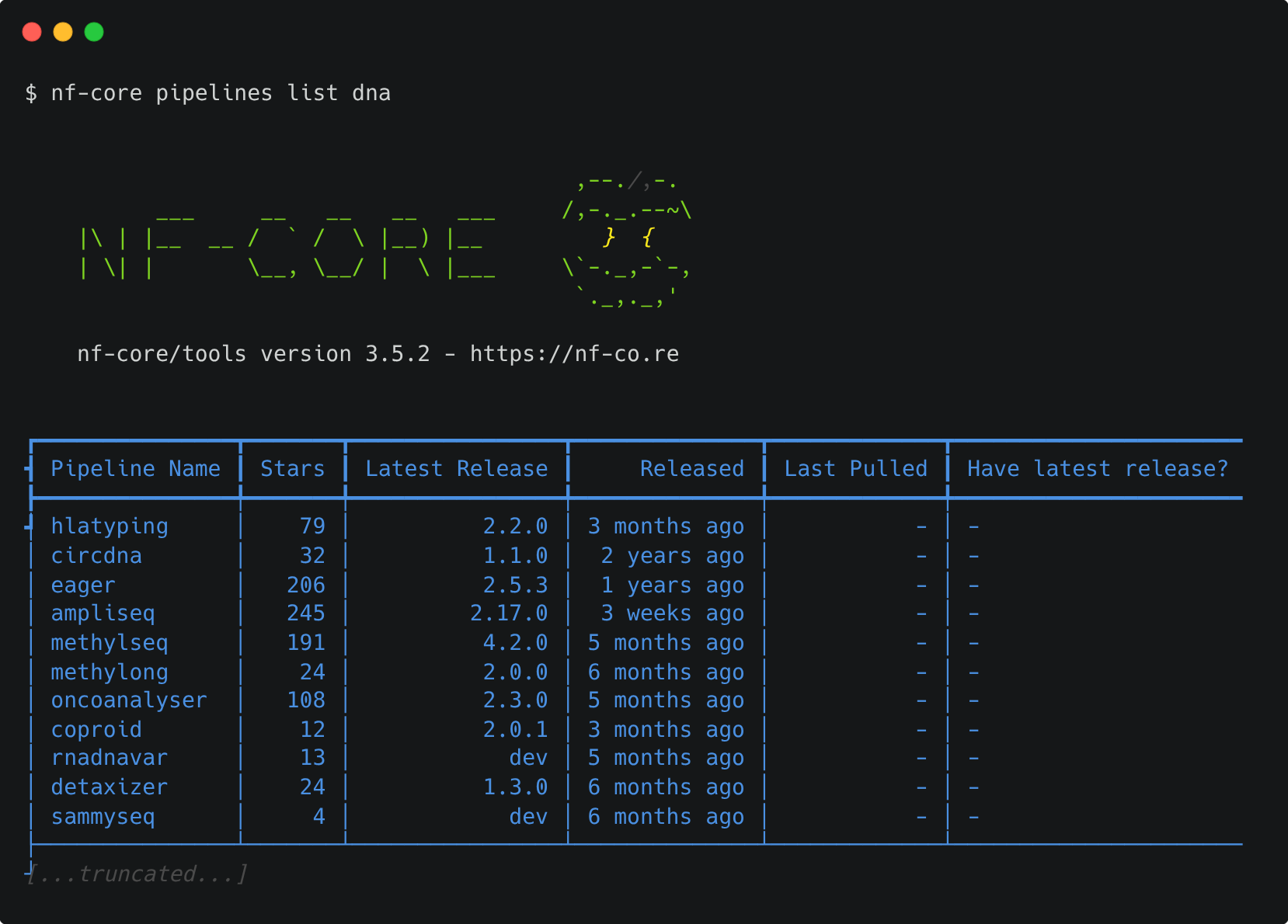
Options can also be used to sort the pipelines by latest release (-s release, default), when you last pulled a pipeline locally (-s pulled), alphabetically (-s name), or number by the number of GitHub stars (-s stars).
nf-core launch
A pipeline can have a large number of optional parameters. To help with this, the nf-core launch command is designed to help you write parameter files for when you launch your pipeline.
The nf-core launch command takes one argument - either the name of an nf-core pipeline which will be pulled automatically or the path to a directory containing a Nextflow pipeline:
When running this command, you will first be asked about which version of the pipeline you would like to execute. Next, you will be given the choice between a web-based graphical interface or an interactive command-line wizard tool to enter the pipeline parameters. Both interfaces show documentation alongside each parameter, will generate a run ID, and will validate your inputs.
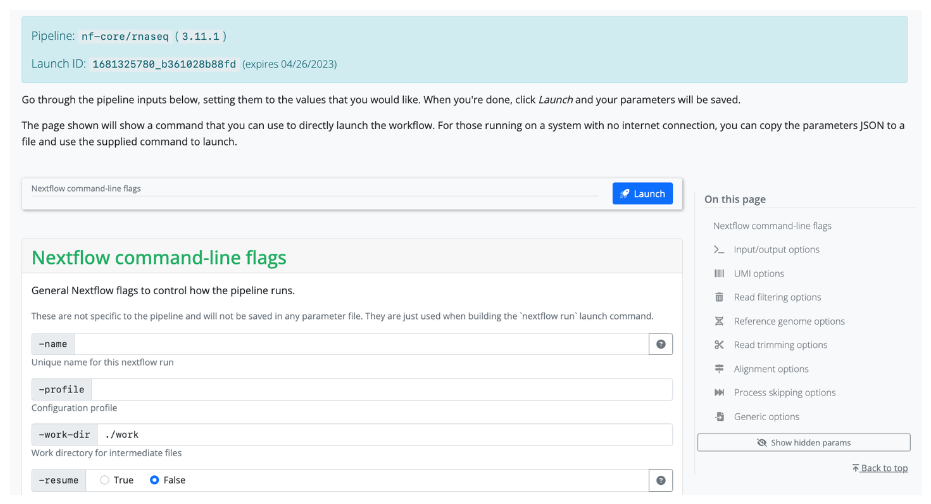
The nf-core launch tool uses the nextflow_schema.json file from a pipeline to give parameter descriptions, defaults, and grouping. If no file for the pipeline is found, one will be automatically generated at runtime.
The launch tool will save your parameter variables as a .json file called nf-params.json. It will also suggest an execution command that includes the -params-file flag and your new nf-params.json file. The command line wizard will finish by asking if you want to launch the pipeline. Any profiles or options that were set using the wizard will be included in your run command.
You can also use the launch command directly from the nf-core launch website. In this case, you can configure your pipeline using the wizard and then copy the outputs to your terminal or use the run id generated by the wizard. You will need to be connected to the internet to use the run id.
Other workshops
This workshop is part of a series of Nextflow workshops we have developed at SIH. For more information on running Nextflow pipelines, see the other workshops in the series:
- Nextflow for Life Sciences: An introduction to writing your own Nextflow pipelines within the context of the life sciences and bioinformatics.
- Nextflow for HPC workshop: A deeper dive into customising and optimising Nextflow pipelines specifically for running on HPC systems.