2.3 Case study: customisation in action
Objectives
- Execute a non-default analysis with an nf-core pipeline
- Troubleshoot pipeline execution failure
- Use custom configuration files to resolve the failure
- Add additional custom configuration files to augment run output
2.3.1 Case study introduction
In this section, no new nf-core or Nextflow concepts will be introduced. Instead, we will apply a number of the customisation strategies we have learnt over the last two days to a theoretical real-world customisation scenario.
We will cover a typical journey experienced when you choose to perform a non-standard analysis, including debugging the dreaded error message, searching for the issue solution, and working out how to make the solution work for you.
We will apply strategic thinking and methodology to:
- Identify and apply changes to the workflow that best suit our experiment goals using pipeline parameters
- Apply the parameters to the run and investigate an error message
- Use multiple custom configurations to resolve the issue
- Customise resource tracing and QC files for tailored analysis reporting
🐭 The scenario
Our mouse gene expression study needs preliminary results for a looming deadline. We have been tasked with customising the nf-core/rnaseq pipeline to identify expressed genes in the dataset in a shorter analysis time.
2.3.2 Devising and running a non-default analysis
Warning
nf-core pipelines are extensively tested, yet with so many tool options and pipeline parameters it would be impractical for every possible combination of workflow choices to be tested. This means that a customised run may at times encounter an error.
This exercise will teach some practical steps to finding an issue solution and applying it using the customisation skills we have covered in the workshop.
While the exact issue described in this lesson is likely to be transient (i.e. fixed in updated nf-core/rnaseq versions), the included troubleshooting and customisation steps can be applied to other issues you may encounter when customising nf-core runs.
Knowing that the longest-running part of the workflow is the STAR_ALIGN process, we check the nf-core/rnaseq user guide and see that the alignment step can be customised to use Hisat2 or Bowtie2 aligner with a pipeline parameter.

Checking out the user guides for these tools, we find Bowtie2 is not suited to our type of input data, and read that Hisat2 is fast with low RAM usage. Given the limited RAM we have available, we decide to test if this customisation can meet our pipeline goals.
Exercise 2.3.2  3 mins
3 mins
- Using the
nf-core/rnaseqparameters guide, find the parameter needed to use the Hisat2 aligner - Add the new parameter to
run_rnaseq.sh - Update the
--outdirvalue tolesson-2.3 - Remove the index parameters for STAR and Salmon since they are no longer required
- Add the Nextflow
-resumeoption - Save the script, then run the updated pipeline by entering the command
bash run_rnaseq.sh
Solution
The parameter needed to use the Hisat2 aligner is:
#!/bin/bash
# parameters
samplesheet=~/data/samplesheet.csv
output_directory=lesson-2.3
ref_fasta=~/data/mm10_reference/mm10_chr18.fa
ref_gtf=~/data/mm10_reference/mm10_chr18.gtf
# configurations
profile=workshop
config=workshop_profile.config
nextflow run rnaseq/main.nf \
--input ${samplesheet} \
--outdir ${output_directory} \
--fasta ${ref_fasta} \
--gtf ${ref_gtf} \
--skip_markduplicates true \
--save_trimmed true \
--save_unaligned true \
--aligner hisat2 \
-profile ${profile} \
-c ${config} \
-resume
2.3.3 Troubleshooting a failed custom pipeline run
If your run completed without error, you can still conduct the troubleshooting steps, using the expected error message displayed above.
Step 1: What does the error say?
It appears to have something to do with /tmp. The samtools error shows that the piped command received no output, so the aligner didn't process any reads. This means our input data is likely not the cause.
Step 2: Is it a filesystem permissions or missing directory issue?
ls -l /tmp shows the directory exists and that we have read and write access to the VM /tmp directory.
Step 3: Does the process command reveal any potential issues?
In Lesson 2.1.3 we learnt that the command executed by a process was saved within the .command.sh file in the process work directory and used the nextflow log command to find this command file.
In this case, we don't need to hunt for the command file as the process work directory has been helpfully printed out alongside the error message.
Note: the work directory below is an example of terminal output, your work directory path will be different
Exercise 2.3.3.1 2 mins
View the .command.sh file within the failed process work directory, or the example below if your run completed successfully.
The command shows no obvious issues, with our sample name, .fq.gz input file and reference genome files correctly populated within the process command. No clues here!
INDEX=`find -L ./ -name "*.1.ht2*" | sed 's/\.1.ht2.*$//'`
hisat2 \
-x $INDEX \
-U SRR3473988_trimmed_trimmed.fq.gz \
\
--known-splicesite-infile mm10_chr18.filtered.splice_sites.txt \
--summary-file SRR3473988.hisat2.summary.log \
--threads 2 \
--rg-id SRR3473988 --rg SM:SRR3473988 \
--un-gz SRR3473988.unmapped.fastq.gz \
--met-stderr --new-summary --dta \
| samtools view -bS -F 4 -F 256 - > SRR3473988.bam
Step 4: Is this a known/previously reported issue for the tool or the nf-core pipeline?
Exercise 2.3.3.2 3 mins
- In a web browser, navigate to the
nf-core/rnaseqgithub repository - Select the 'Issues' tab, and search for issues matching the term 'hisat2'
- Also search for issues relating to tmp/temp directory errors in the Hisat2 github repository
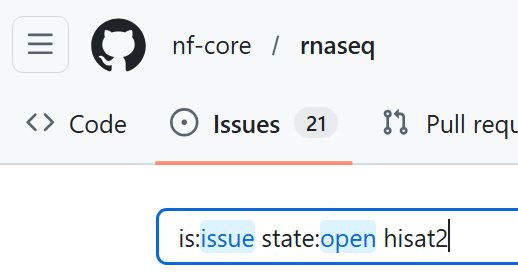
Can you find any issues relating to the (ERR): mkfifo(/tmp/45.unp) error in either of these repositories?
Solution
The nf-core/rnaseq issues search was not revealing, however a Hisat2 issue titled Enabling a --temp-directory parameter #438 describes the exact error message some of use have encountered.
Reading through the issue activity, you can see that the issue has been linked by an nf-core issue titled update module: HISAT2/ALIGN #9487.
Following this link, we can see that nf-core have marked this issue as 'WIP' (work in progress). Seems like we have found a solution to our bug!
The importance of open-source and community contribution
By reporting issues and bugs we find in open-source code through github issues, we can effect real change. Developers are typically welcoming of bug reports that help improve their code and happy to help you to use their tool for your research. Other users of a tool may also chime in with their solutions, or you may want to add your helpful tips to an open issue!
2.3.4 Configuring the solution
The ticket describes a new release of Hisat2 v 2.2.2 that features a new parameter to prevent the /tmp error.
To customise this version of the nf-core/rnaseq pipeline to use Hisat2, we will:
- Over-ride the default version of Hisat2 to use the latest version
- Add the new parameter to the Hisat2 process command
2.3.4.1 Running with multiple custom configuration files
Nextflow allows you to add as many configs as needed to customise a run.
These can be supplied to the Nextflow parameter -c in a comma-delimited list, for example:
Alternatively, -c can be included several times, for example:
Custom config order matters!
Recall from Lesson 1.3.7 that order matters, with the right-most custom config files taking precedence.
In the examples above, a configuration in custom-2.config would over-ride custom-1.config if both configured the same attribute.
Some configurations make sense being grouped together within a single config file, for example customising compute resources and software management within an institutional config.
Others are best placed within individual and explicitly named configs to ensure they are only applied when needed (i.e., they are modular).
As we learnt in Lesson 2.2.4.2, we can also group configuratons that apply to a certain environment or analysis within a custom profile. Once we have tested and verified our Hisat2 customisation is working, we will incorporate it into our workshop custom profile.
2.3.4.2 Applying a custom tool version
Recall from Lesson 2.1.6.3 that all tool versions are included within the MultiQC report as well as a yaml file in the pipeline_info folder.
Exercise 2.3.4.2.1 3 mins
Discover the version of Hisat2 used in our most recent run of nf-core/rnaseq.
Hint: Failed run?
If your run failed, the MultiQC report was not generated! Use the pipeline_info/nf_core_rnaseq_software_mqc_versions.yml file, which will include versions for the modules that successfully completed before the failure point.
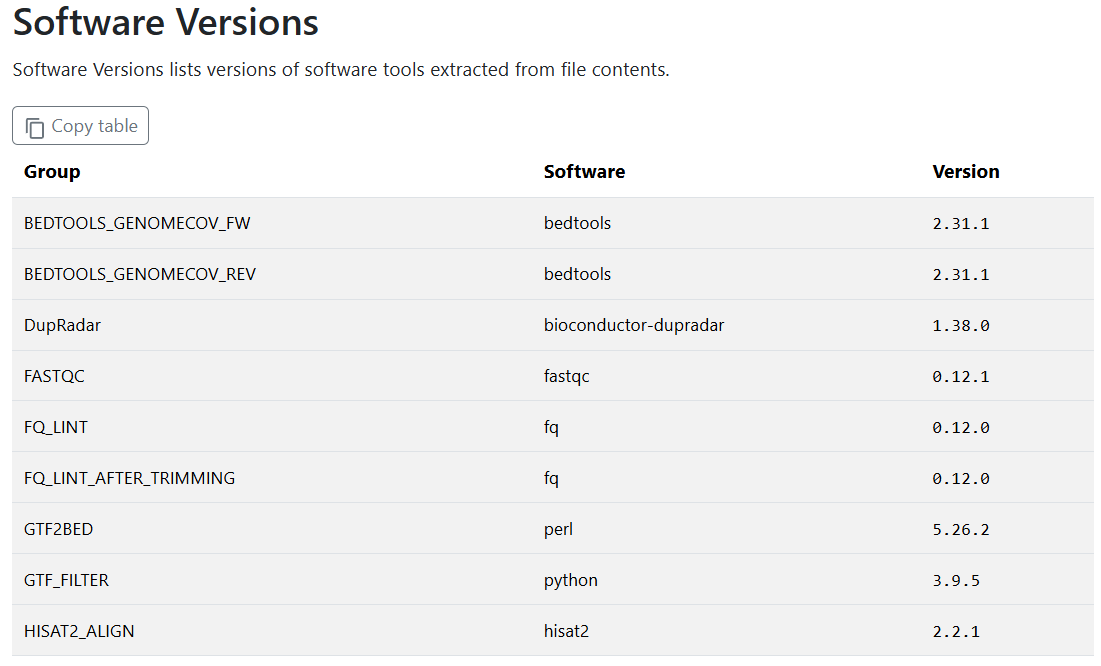
We need Hisat2 version 2.2.2 for the new --temp-dir parameter. How should we apply this customisation to our our run?
Poll 2.3.4.2 3 mins
For maintaining portability and reproducibility, what is the optimal method to apply our custom Hisat2 container to our pipeline run?
a) Within the nectar_vm.config institutional config file. It's already working well on our platform, so we can build on it for further customisations without issue.
b) Within a separate custom config file named custom-hisat2.config. We would then add it to any runs of nf-core/rnaseq that required this non-default workflow.
c) Within the Hisat2 module main.nf file. This way, we don't need to worry about adding another custom config when we execute the run command.
Answer
b) Within a separate custom config file named custom-hisat2.config. This method is the most explicit, modular, portable, and reproducible.
By placing it within a clearly named custom config file, there is less chance of unwittingly executing a workflow that does not match the expected utilisation of tools. Making it a separate config also means it can be easily dropped from our run when nf-core finalises their testing of the issue marked as 'WIP' and includes the resolution within the next version of rnaseq.
Institutional configs are intended to be shareable with others using nf-core pipelines on the same infrastructure. Adding the custom tool over-ride to this file would prevent our institutional config from being portable to other analyses, so a) is incorrect.
In general, editing the process main.nf file is not recommended, as this impedes reproducibility. Swapping out tool versions is a bespoke adaptation of the nf-core workflow that would harm reproducibility if it was inadvertently executed, which is likely to happen if it is hidden within the source code, making c) incorrect.
To make the purpose of the config clear to anyone else in the lab who runs the pipeline, we will also include a descriptive comment explaining why the config is needed.
Comments in Nextflow
Commenting code is good practice to make the purpose and logic of the code easier for others and your future self to understand.
Comments in Nextflow use // for single line comments or /* <comment lines> */ to comment multiple lines.
Exercise 2.3.4.2.2 5 mins
- Create a new file called
custom-hisat2.config - Add a Nextflow comment describing what the config does and why
- Identify the fully qualified process name/process execution path for the
HISAT2_ALIGNprocess - Within a
processscope code block, provide the process name identified above to the appropriate process selector - Within the process selector code block, add the path to the Hisat2 v 2.2.2 container described in the nf-core issue
Hint: Process execution paths
You can obtain the process execution path from the execution trace, timeline or report files within <outdir>/pipeline_info.
Process names are not always unique in nf-core pipelines that may contain modules from other sources. Best practice for specificity is to use the fully qualified process name, which typically takes the form:
PIPELINE_NAME:WORKFLOW_NAME:SUBWORKFLOW_NAME:PROCESS_NAME
Hint: Process selectors
We learnt about process selectors in Lesson 2.2.5.1.
Hint: Syntax help
Our nectar_vm.config files contain process scope and process selector code blocks. Our hisat2 config will be more simple, not requiring the profile or singularity scopes we have within our Nectar institutional config.
Hint: Hisat2 v 2.2.2 container path
This was included in the nf-core issue:
Solution
// Custom config to apply latest Hisat2 v 2.2.2 to nf-core/rnaseq v 3.23.0
// Required to add new Hisat2 --temp-directory parameter to resolve known issue
// Hisat2 issue: https://github.com/DaehwanKimLab/hisat2/issues/438
// nf-core issue: https://github.com/nf-core/modules/issues/9487
//
// This config should be dropped when running future nf-core/rnaseq versions that resolve the issue
process {
withName: "NFCORE_RNASEQ:RNASEQ:FASTQ_ALIGN_HISAT2:HISAT2_ALIGN" {
container = 'https://community-cr-prod.seqera.io/docker/registry/v2/blobs/sha256/c3/c36472269e8898f63b7b65dd40433462d541f9e75f9401f0bf8488021275d006/data'
}
}
Use caution when customising tool versions
Note that for deploying nf-core pipelines, it is generally not recommended to change the tool versions, as this will decrease portability and reproducibilty as well as being an untested pipeline.
This exercise is to demonstrate how you can specify containers, as this may aid you in developing and testing your own Nextflow workflows or testing new tool versions. When a non-standard version of a tool is used for publishing nf-core pipeline results, this should be clearly stated within the methods section of the publication.
2.3.4.3 Adding a custom tool parameter
As introduced in Lesson 1.3.8, nf-core modules define an ext.args process directive that can be used to add any tool parameter to a process command.
The utility of this directive is wide-reaching when you consider the number of optional parameters a typical bioinformatics tool may have. It is not feasible for nf-core to parameterise all of these arguments...
Extra convenience has been added for some commonly-used tools by wrapping the ext.args directive within an nf-core pipeline parameter, typically named --extra_<tool>_args.
Exercise 2.3.4.3.1 2 mins
- Open the
nf-core/rnaseqparameters online guide and check for any--extra_<tool>_argsparameters - Can you find a pipeline parameter to add extra arguments to the Hisat2 tool?
Solution
There is no such parameter for Hisat2.
Included in version 3.23.0 of nf-core/rnaseq are:
--extra_trimgalore_args--extra_fastp_args--extra_star_align_args--extra_bowtie2_align_args--extra_salmon_quant_args--extra_kallisto_quant_args--extra_fqlint_args
The ext.args directive typically forms part of all nf-core module code, even if the tool itself does not have an extra_<tool>_args parameter. This provision enables you to add any tool parameter to any nf-core pipeline tool with a custom configuration file that passes your parameter to the target module via ext.args.
Let's see this in action!
Exercise 2.3.4.3.2 3 mins
- Using the procedure described in Exercise 2.2.2.3, find the
main.nffile for the HISAT2_ALIGN process:
The nf-core subworkflow named FASTQ_ALIGN_HISAT2 shows the module path for HISAT2_ALIGN as modules/nf-core/hisat2/align/main.nf.
- Open the file with
code rnaseq/modules/nf-core/hisat2/align/main.nffile - View the file to observe the Nextflow code that defines a variable called
argsand assigns it the contents ofext.args
If ext.args is empty (i.e. the run does not add any extra arguments to this tool), the args variable remains empty in the process script. If it has been provided to the run, $args will be expanded to the supplied custom argument in the process script:
26 script:
27 def args = task.ext.args ?: ''
Further down, within the hisat2 command that is executed by the process, we can see the $args variable added to the hisat2 command:
42 hisat2 \\
43 -x \$INDEX \\
44 -U $reads \\
45 $strandedness \\
46 $ss \\
47 --summary-file ${prefix}.hisat2.summary.log \\
48 --threads $task.cpus \\
49 $rg \\
50 $unaligned \\
51 $args \\
52 | samtools view -bS -F 4 -F 256 - > ${prefix}.bam
Now that we have verified that the pipeline soure code can accept our argument via ext.args, we will add it to our custom-hisat2.config file created in the previous lesson, and run our updated pipeline with multiple custom configuration files.
Exercise 2.3.4.3.3 3 mins
- Provide
--temp-directory .to theext.argsdirective within the process selector code block ofcustom-hisat2-version.config - Update
run_rnaseq.shby addingcustom-hisat2.configto the Nextflow-cparameter, and ensure the Nextflow-resumeflag is applied - Change
--outdirtolesson-2.3.4 - Save both scripts, then run the updated pipeline by entering the command
bash run_rnaseq.sh
Paths inside containers
When running tools with containers, the process is running inside the container, so the . path will evaluate to a location inside the container.
In our case, this will ensure that the tmp directory used by each process will be unique, and avoid error-inducing clashes when multiple HISAT2_ALIGN processes attempt to write to the user's /tmp on the local machine.
Solution
// Custom config to apply latest Hisat2 v 2.2.2 to nf-core/rnaseq v 3.23.0
// Required to add new Hisat2 --temp-directory parameter to resolve known issue
// Hisat2 issue: https://github.com/DaehwanKimLab/hisat2/issues/438
// nf-core issue: https://github.com/nf-core/modules/issues/9487
//
// This config should be dropped when running future nf-core/rnaseq versions that resolve the issue
process {
withName: "NFCORE_RNASEQ:RNASEQ:FASTQ_ALIGN_HISAT2:HISAT2_ALIGN" {
container = 'https://community-cr-prod.seqera.io/docker/registry/v2/blobs/sha256/c3/c36472269e8898f63b7b65dd40433462d541f9e75f9401f0bf8488021275d006/data'
ext.args = '--temp-directory .'
}
}
#!/bin/bash
# parameters
samplesheet=~/data/samplesheet.csv
output_directory=lesson-2.3.4
ref_fasta=~/data/mm10_reference/mm10_chr18.fa
ref_gtf=~/data/mm10_reference/mm10_chr18.gtf
# configurations
profile=workshop
config=workshop_profile.config
hisat2=custom-hisat2.config
nextflow run rnaseq/main.nf \
--input ${samplesheet} \
--outdir ${output_directory} \
--fasta ${ref_fasta} \
--gtf ${ref_gtf} \
--skip_markduplicates true \
--save_trimmed true \
--save_unaligned true \
--aligner hisat2 \
-profile ${profile} \
-c ${config},${hisat2} \
-resume
🤞 Hopefully your run now completes without error!
2.3.4.5 Verifying customisations
In some cases, the successful application of customisations to a run are self-evident in the pipeline outputs. In other cases, they may not be, and you may want to perform verification.
In this scenario, the error only occurs if more than one Hisat2 process runs at the same time, which is why your initial Hisat2 run may or may not have encountered an error.
Exercise 2.3.4.5.1 2 mins
Check that the custom version 2.2.2 of hisat2 was run.
Hint
Recall from Lesson 2.1.6.3 that all tool versions are included within the MultiQC report as well as a yaml file in the pipeline_info folder.
Solution
By viewing the Software Versions heading in the MultiQC report:
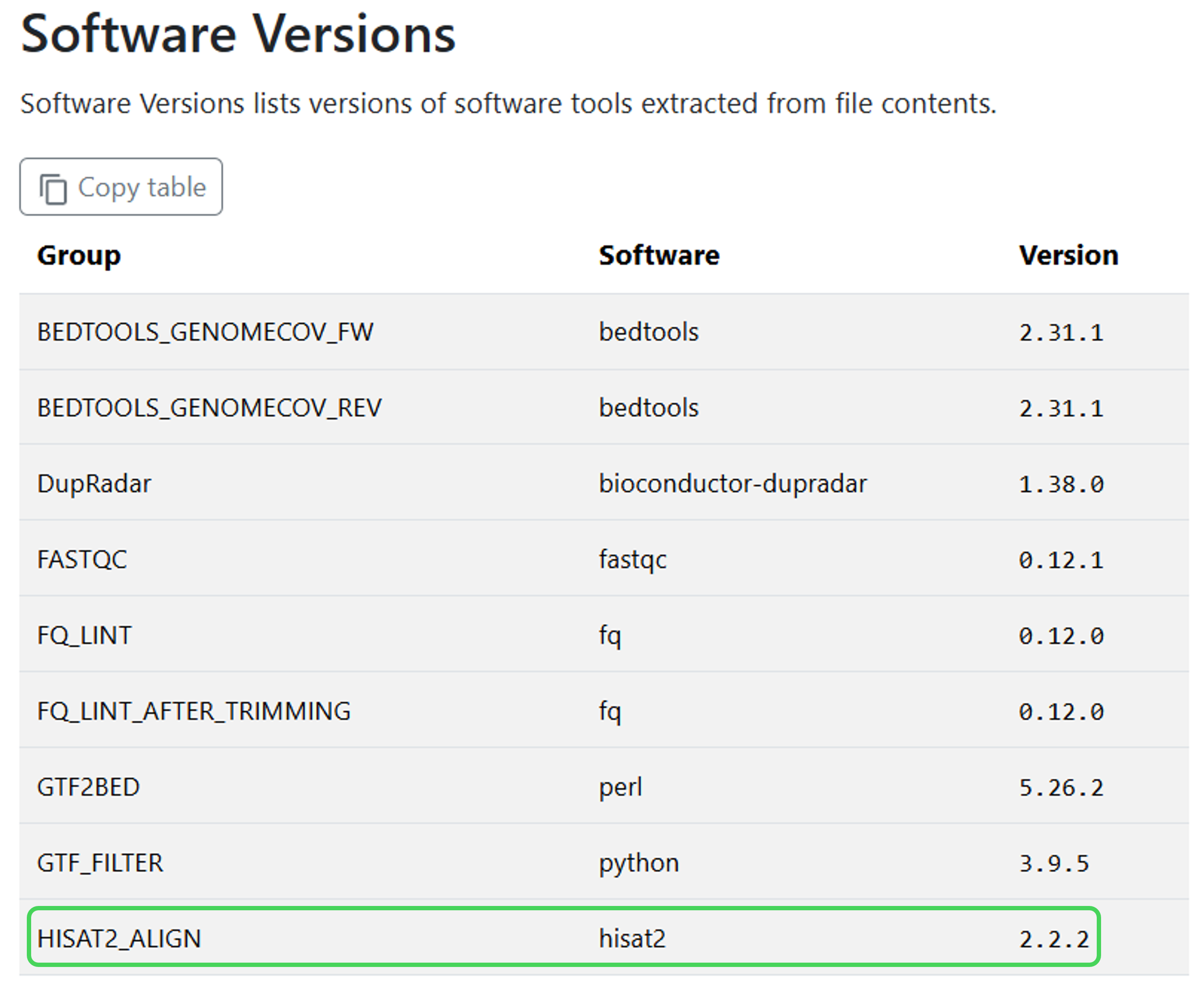
Or using the yaml file in the pipeline_info folder:
Exercise 2.3.4.5.2 5 mins
- Check that the custom parameter
--temp-directory .was applied to the HISAT2_ALIGN process command
Hint
Finding the .command.sh for a process within the process work directory using nextflow log was practised in Exercise 2.1.3.
Solution
- Check that the custom parameter
--temp-directory .was applied to the HISAT2_ALIGN process command:
hisat2 \
-x $INDEX \
-U SRR3473989_trimmed_trimmed.fq.gz \
\
--known-splicesite-infile mm10_chr18.filtered.splice_sites.txt \
--summary-file SRR3473989.hisat2.summary.log \
--threads 2 \
--rg-id SRR3473989 --rg SM:SRR3473989 \
--un-gz SRR3473989.unmapped.fastq.gz \
--temp-directory . \
| samtools view -bS -F 4 -F 256 - > SRR3473989.bam
Or using an alternate command:
NFCORE_RNASEQ:RNASEQ:FASTQ_ALIGN_HISAT2:HISAT2_ALIGN (SRR3473989)
INDEX=`find -L ./ -name "*.1.ht2*" | sed 's/\.1.ht2.*$//'`
hisat2 \
-x $INDEX \
-U SRR3473989_trimmed_trimmed.fq.gz \
\
--known-splicesite-infile mm10_chr18.filtered.splice_sites.txt \
--summary-file SRR3473989.hisat2.summary.log \
--threads 2 \
--rg-id SRR3473989 --rg SM:SRR3473989 \
--un-gz SRR3473989.unmapped.fastq.gz \
--temp-directory . \
| samtools view -bS -F 4 -F 256 - > SRR3473989.bam
👍 HISAT2_ALIGN is using the custom tool version and custom parameter to resolve our issue.
2.3.4.6 Selecting processes with regular expressions
HISAT2_ALIGN has used our custom container version, but not the other Hisat2 modules within the rnaseq pipeline. Version consistency within data analysis is crucial for preventing potential version conflicts in execution or outputs, as well as reproducibility.
To apply a custom container version to all processes in the pipeline that utilise a given tool, we can use a regular expression with withName. This is possible due to the standardised way that nf-core names its processes, where the software/tool name is typically included in the process name.
Exercise 2.3.4.6 7 mins
- Within
custom-hisat2.config, create a newwithNamecode block and use the.*Nextflow wildcard-style regex to match all processes with 'HISAT2' in the name - Move the custom container line to the new
withNamecode block - Save the config, then run the updated pipeline with
bash run_rnaseq.sh
Nextflow pattern match syntax
A pattern match (or 'regular expresson') in Nextflow uses forward slashes around the pattern, i.e. /<pattern-to-match>/.
A wildcard-style pattern uses .* to match any character any number of times.
Solution
- Check the tool version for other Hisat2 processes (HISAT2_BUILD, HISAT2_EXTRACTSPLICESITES) using either of the methods shown in Exercise 2.3.4.5.1
2.3.5 Customising resource tracing
As observed in Lesson 2.2.5.2, the pipeline_info execution report shows resources used in graphical form. A text file that is easily parsed for example using R or Python can be more flexible in meeting custom reporting needs, for example making plots of resource usage.
The nf-core pipeline_info trace file, printed to the pipeline output directory by default, is a plain tab-delimited file that contains key details about processes. There are a number of other available fields, and the trace file can be customised to include any combination of fields using a custom configuration file.
The rnaseq/nextflow.config file enables the trace option and sets a unqiue filename using date and timestamp:
140 trace_report_suffix = new java.util.Date().format( 'yyyy-MM-dd_HH-mm-ss')
362 trace {
363 enabled = true
364 file = "${params.outdir}/pipeline_info/execution_trace_${params.trace_report_suffix}.txt"
365 }
Note that the trace.fields option is absent, meaning nf-core is using the default fields for Nextflow trace. To discover the default fields when trace is enabled, view the fields included in the example trace file, or the pipeline_info/execution_trace_<date>_<time>.txt from one of your completed runs from this session.
There is no trace option that allows adding fields to the default, but we can over-ride the default fields with our custom fields using the trace.fields option within a custom configuration file.
Nextflow log fields vs trace fields
Recall the nextflow log -l command introduced in Lesson 1.2.5.
The fields printed by nextflow log -l and their meanings are identical to the fields available to trace.fields with an important exception. Since the % symbol is a special character, %cpu and %mem as described for Nextflow trace correspond to pcpu and pmem for the nextflow log -f command-line utility.
Exercise 2.3.5.1 5 mins
To assess whether our customisations have met the goal of increased analysis speed, we would like to plot run time and compute usage of our custom Hisat2 pipeline to the default STAR pipeline.
For our plots, we need to gather per-process metrics on walltime, CPUs requested and percentage used, memory requested and percentage used.
- View the available trace fields and identify the names of the fields we need
- Check which fields are included in the default trace file and identify any from our list that are not included
- Create a new custom configuration file called
custom-trace.config, add a descriptive comment, and add atracescope code block - Within the
tracescope, provide a comma-separated list of fields to include to thefieldsoption of trace- You can choose whether to list all default fields plus our custom fields, or a shorter list including just the fields we want to plot
- If you choose a shorter list of fields, include the
namefield to identify which processes the resource metrics apply to - Your list should be enclosed in quotes, eg
fields = 'field1,field2'
Solution
The field names we need are:
- `cpus`
- `%cpu`
- `memory`
- `%mem`
- `realtime`
Of these, the following are included in the nf-core/rnaseq trace file by default:
- `%cpu`
- `realtime`
The custom fields we need to add are:
- `cpus`
- `memory`
- `%mem`
The next step is to add the new config to our run. The -c config list is growing rather long:
Each config is a discrete set of customisations matching a specific purpose - they are modular and portable - yet each of them are required for the specific application of running this non-standard run on these VMs. It makes sense then to logically group these configs under our workshop profile.
Exercise 2.3.5.2 5 mins
- Open
workshop_profile.config - Under the
workshopprofile, addcustom-trace.configusing theincludeConfigoption - Now we have tested and verified the new Hisat2 pipeline and issue-solving config, it makes sense to group it under the profile
- Add
custom-hisat2.configto theworkshopprofile using theincludeConfigoption - Update
run_rnaseq.shby removingcustom-hisat2.configfrom the Nextflow-cparameter
- Add
- Within
run_rnaseq.sh, change--outdirtolesson-2.3.5 - Save both scripts, then run the updated pipeline by entering the command
bash run_rnaseq.sh
Solution
profiles {
workshop {
includeConfig 'nectar_vm.config'
includeConfig 'custom-trace.config'
includeConfig 'custom-hisat2.config'
}
}
#!/bin/bash
# parameters
samplesheet=~/data/samplesheet.csv
output_directory=lesson-2.3.5
ref_fasta=~/data/mm10_reference/mm10_chr18.fa
ref_gtf=~/data/mm10_reference/mm10_chr18.gtf
# configurations
profile=workshop
config=workshop_profile.config
nextflow run rnaseq/main.nf \
--input ${samplesheet} \
--outdir ${output_directory} \
--fasta ${ref_fasta} \
--gtf ${ref_gtf} \
--skip_markduplicates true \
--save_trimmed true \
--save_unaligned true \
-profile ${profile} \
-c ${config} \
-resume
Does the new resource trace file meet our plotting needs?
Exercise 2.3.5.3 1 min
- Open the file
lesson-2.3.5/pipeline_info/execution_trace_<date>_<time>.txtby double clicking it in the VS Code explorer pane - Check that the custom fields you have specified have been applied
Tracing cached processes
For processes with status CACHED, the resources reported in the trace file (as well as other pipeline_info files) are from the actual compute required to execute the process, and not the time this execution took to restore the data from cache. Keeping all the usage details together whether or not tasks were cached simplifies reviewing and reporting compute usage.
Warning
When you launch a run with custom configuration files, these will be printed in the launch log as your run starts, as well as documented in the Nextflow command section of the Nextflow workflow report.
Configuration files that are included within a profile are not listed in this way, only the profile itself is listed.
This makes it crucial that you save your custom configuration files along with with pipeline outputs to ensure reproducibility and portability of your custom analysis.
2.3.6 Personalise MultiQC reports
Many nf-core pipelines use MultiQC to aggregate results and statistics output by various bioinformatics tools, helping to summarise experiments containing multiple samples and multiple analysis steps. Recall that we used the MultiQC report to debug the strandedness error in Lesson 2.1.4.
MultiQC reports can be customised on the MultiQC command line or within a yaml-formatted MultiQC configuration file. nf-core has enabled this functionality with the --multiqc_config parameter, which takes the yaml-formatted MultiQC configuration file and applies all included MultiQC parameters to the MultiQC process command.
Hidden nf-core parameters
The --multiqc_config parameter is a hidden parameter! To see this parameter, you need to apply --showHidden on the command line help, or select 'Show hidden' on the lower right hand side of the nf-core parameters webpage.
To polish up our presentation at the next mouse project meeting, we want to add some custom detail to the MultiQC report. We will create a custom MultiQC config to:
- Add a custom report header and title
- Overlay a theoretical GC content track specific for our mouse reference genome
These are just two examples of the many MultiQC configurations available.
Exercise 2.3.6.1 3 mins
- Create a new file named
custom-multiqc.yaml - Under the
report_header_infoconfig option, add a custom report header- You can add any details you choose under
report_header_info, as long as they conform to the yaml key:value pairs format
- You can add any details you choose under
- Under the
fastqc_configoption, specifyfastqc_theoretical_gcas the yaml key andmm10_txome(mouse transcriptome) as the yaml value - Give your MultiQC report file a descriptive filename using the
filenameconfig option - Add some titles and introductory text if desired
- Save the file
Solution
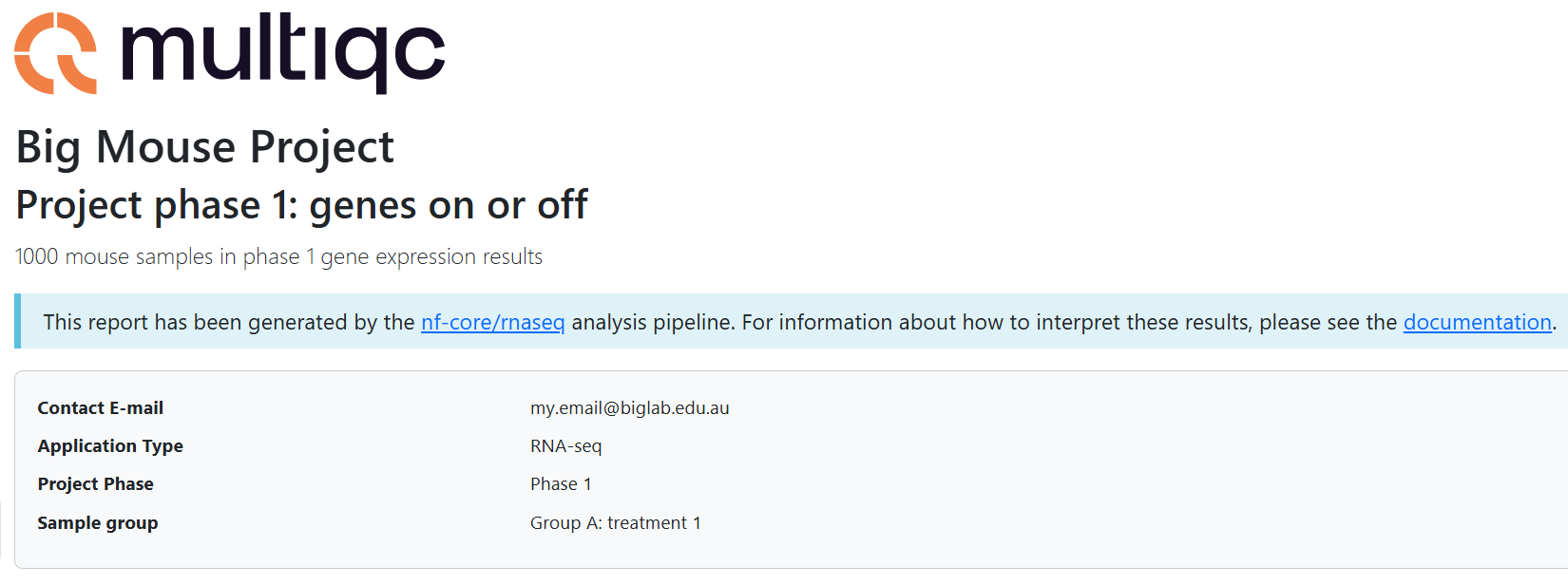
title: "Big Mouse Project"
filename: Big_Mouse_Project_phase1_groupA_rnaseq_QCreport.html
subtitle: "Project phase 1: genes on or off"
intro_text: "1000 mouse samples in phase 1 gene expression results"
report_header_info:
- Contact E-mail: "my.email@biglab.edu.au"
- Application Type: "RNA-seq"
- Project Phase: "Phase 1"
- Sample group: "Group A: treated"
fastqc_config:
fastqc_theoretical_gc: "mm10_txome"
You may be expecting to add this configuration file at the Nextflow -c parameter in your run command. In this case, we do not add the configuration at -c!
This configuration is a yaml-formatted file matching the input format expected by the tool MultiQC, and not a Nextflow configuration file. nf-core/rnaseq includes the pipeline parameter --multiqc_config (select 'Show hidden' to find this parameter!) so we can add this particular configuration as we would any other pipeline parameter.
Exercise 2.3.6.2 3 mins
- Add the MultiQC yaml file to your run command in
run_rnaseq.sh - Update the
--outdirto lesson-2.3.6 - Save your run script, then run the updated pipeline by entering the command
bash run_rnaseq.sh
Solution
#!/bin/bash
# parameters
samplesheet=~/data/samplesheet.csv
output_directory=lesson-2.3.6
ref_fasta=~/data/mm10_reference/mm10_chr18.fa
ref_gtf=~/data/mm10_reference/mm10_chr18.gtf
# custom multiqc
multiqc=custom-multiqc.yaml
# configurations
profile=workshop
config=workshop_profile.config
nextflow run rnaseq/main.nf \
--input ${samplesheet} \
--outdir ${output_directory} \
--fasta ${ref_fasta} \
--gtf ${ref_gtf} \
--skip_markduplicates true \
--save_trimmed true \
--save_unaligned true \
--multiqc_config ${multiqc} \
-profile ${profile} \
-c ${config} \
-resume
Does the MultiQC report have the intended customisations?
Exercise 2.3.6.3 3 mins
- Once your run has completed, right-click on
lesson-2.3.6/multiqc/hisat2/<custom-title>.htmland select 'Open with Live Server'- Notice your customisations in the header section of the report, for example:
- Navigate to the section titled FastQC: Per Sequence GC Content and check that the custom track has been successfully added
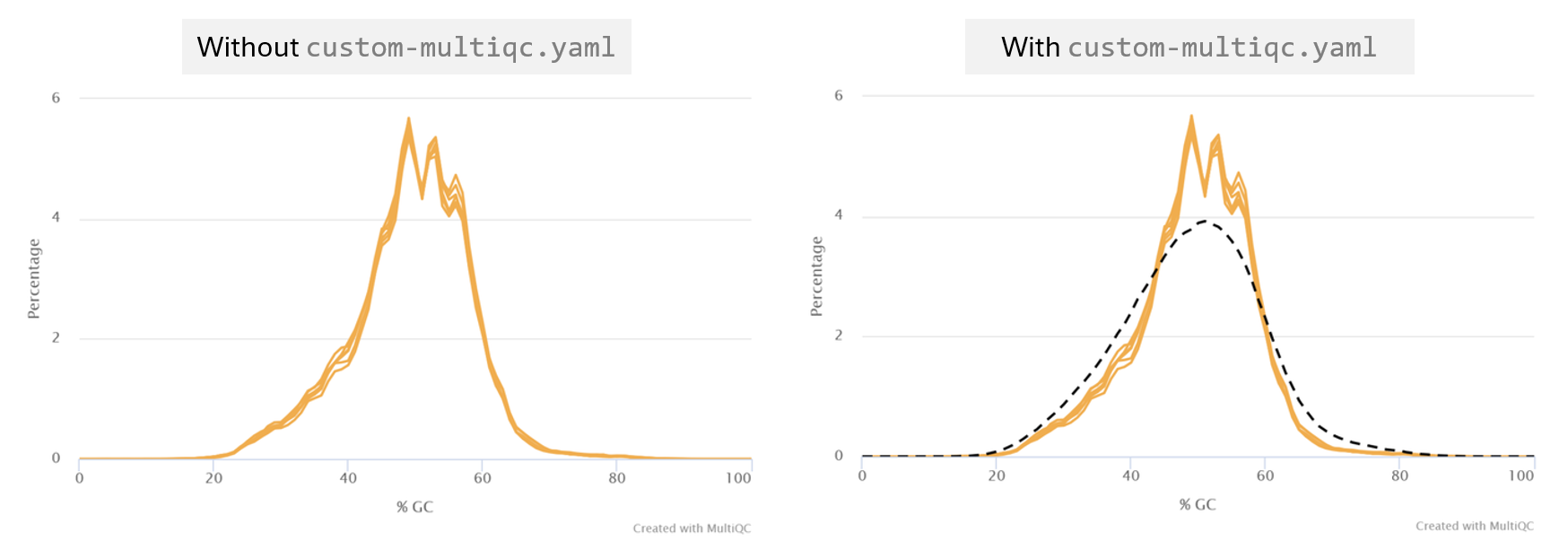
 The custom GC track supports that our sample transcriptomes follow a normal distribution consistent with the mm10 reference.
The custom GC track supports that our sample transcriptomes follow a normal distribution consistent with the mm10 reference.
💃🏽🕺🏻 The mouse project data is analysed in time to meet the deadline and our colleagues are impressed by the fancy MultiQC report!
2.3.7 Workshop key points
Key points from Session 1 and Session 2
- nf-core is a curated set of analysis workflows built using the workflow language Nextflow
- There are active community platforms you can join for nf-core and Nextflow to ask questions and stay up to date with the latest activities and announcements
- Understanding how to customise nf-core workflows through pipeline parameters and configuration files enables you to adapt nf-core pipelines to real-world analysis situations
- Understanding the priority order in which parameters and configurations are applied helps you ensure your runs receive the instructions intended
- Understanding Nextflow configuration and nf-core code structure can help you troubleshoot a run
- Customisations to what is run are controlled by nf-core pipeline parameters as well as configurations targeting specific processes using the
processscope and process selectors - Custom tool parameters that are not covered by an nf-core pipeline parameter can be applied to a run using
ext.argswithin theprocessscope - Customisations to how the run executes on your machine are controlled by custom configuration files
- Multiple configurations can be applied to a run using
-cor grouped into customprofiles - Appropriate modular and descriptively named custom config use maintains portability and reproducibility when customising nf-core workflows
- All configs, parameter files and run scripts should be backed up alongside pipeline results
 nf-core pipelines have comprehensive documentation describing usage, parameters, outputs, and interpreting results. Always consult the docs before running and customising a pipeline!
nf-core pipelines have comprehensive documentation describing usage, parameters, outputs, and interpreting results. Always consult the docs before running and customising a pipeline!