1.3 Configuring nf-core pipelines
Objectives
- Learn how to customize the execution of an nf-core pipeline
- Customize a toy example of an nf-core pipeline
1.3.1 Introduction to Nextflow configuration
There are two ways in which we can control how nf-core and other Nextflow pipelines run: through Nextflow configuration files and pipeline parameters. Configuration files are special .config files which are used to set a wide variety of options that control how the Nextflow engine runs, such as defining computing environments, specifying containerisation systems, and setting resources required by processes. Pipeline parameters, on the other hand, are specific to the pipeline you are running and are used to pass information to the pipeline such as sample information, filtering thresholds, reference files, etc.
1.3.2 Nextflow configuration files
When a pipeline is launched, Nextflow will look for configuration files in several locations. As each configuration file can contain conflicting settings, the sources are ranked to decide which settings to apply. Configuration sources are reported below and listed in order of increasing priority:
$NXF_HOME/config(defaults to$HOME/.nextflow/config)nextflow.configin the project directorynextflow.configin the launch directory- Config files specified with
-c <config-files>
All nf-core pipelines come with a nextflow.config file in their project directory which contains the default configuration options for the pipeline. The other files listed above are all instances of custom configuration files that can be supplied by the user to override the pipeline defaults. Understanding how and when these files are interpreted by Nextflow is critical for the accurate configuration of a pipelines execution.
Project and launch directories
Note how Nextflow looks for two different files, each called nextflow.config, first in the project directory, then in the launch directory. There is a subtle but important difference between these two directories. The project directory is the folder containing the pipeline code, i.e. what you download when running nextflow clone or nf-core pipelines download. The launch directory is your current working directory in which you run nextflow run.
For example, in Lesson 1.2.4 when we downloaded and ran nextflow-io/hello, our launch directory was ~/session1, while the project directory was ~/session1/hello. Were we to create the file ~/session1/nextflow.config and set configuration options inside it, those options would take precedence over any conflicting settings defined in ~/session1/hello/nextflow.config.
This system allows you to finely tune how Nextflow runs for you. An nf-core pipeline will come with its own nextflow.config file, but you can use your own nextflow.config file in your launch directory to override the defaults. You may also have a config file at $HOME/.nextflow/config to set configuration options that you frequently use across different pipelines.
Caveat
While there is a lot of flexibility built in to this configuration system, it can also cause subtle problems. Both the launch directory nextflow.config file and the $NXF_HOME/config files are loaded implicitly, meaning you don't need to tell Nextflow to load them. This can be convenient, but if you forget these files exist when running a new pipeline, you may find that Nextflow behaves unexpectedly!
In general, we recommend that all custom configuration options be passed to Nextflow explicitly using the -c option.
Notably, configuration files can define of one or more profiles. A profile is a set of configuration attributes that are grouped together and can be activated when launching a pipeline by using the -profile command option:
nf-core pipelines define several profiles in their nextflow.config files, which primarily fall under one of three types:
- Software management profiles
- Profiles for specifying the software management tool to use when running the pipeline, e.g.,
docker,singularity, andconda
- Profiles for specifying the software management tool to use when running the pipeline, e.g.,
- Test profiles
- Profiles for executing the pipeline with a standardized set of test data and parameters, e.g.,
testandtest_full - Test profiles are typically used during development to check whether new changes work as expected; they are also a great place to start when learning how to use a new pipeline
- Profiles for executing the pipeline with a standardized set of test data and parameters, e.g.,
- Institutional configuration profiles
- Profiles that load one of the many nf-core institutional configuration files
- These include profiles for for both NCI's Gadi and Pawsey's Setonix HPC clusters
Importantly, multiple profiles can be activated at the same time by specifying them in a comma-separated (,) list when you execute your command. The order of profiles is important as they will be read from left to right. If there is any overlap in the configuration options set by each profile, the right-most profile will take precedence.
Exercise 1.3.2.1  2 mins
2 mins
We have created a small demo pipeline using the nf-core template called nf-core-demo. It's located in a repository on the Sydney-Informatics-Hub GitHub organisation. Over the next several exercises, we're going to use it to see how we can configure how an nf-core pipeline runs.
Start by using nextflow clone to download the pipeline repository to your working directory.
You should get the following message:
If you list the repository contents, you should see the typical directory structure of an nf-core pipeline:
Check your Singularity cache dir is set
For the remainder of the workshop, we are using Singularity containers to manage software installation. This means the tools we use don't need to be installed directly on the machines and we can control exactly which version of each tool we use. For this to work properly, we need to ensure that the NXF_SINGULARITY_CACHEDIR environment variable is set.
Recall in the previous lesson we set this variable in our ~/.bashrc file. Confirm the variable is still set:
You should see the path we chose printed to the terminal:
Exercise 1.3.2.2 3 mins
Now that you have cloned the Sydney-Informatics-Hub/nf-core-demo pipeline, run it with the following profiles:
- Use the
testprofile to automatically provide a test dataset as input - Use the
singularityprofile to tell Nextflow to use Singularity containers to run each of the pipeline modules
N E X T F L O W ~ version 25.10.4
Launching `nf-core-demo/main.nf` [sleepy_mendel] DSL2 - revision: 282d7ef11e
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo v1.0dev
------------------------------------------------------
Core Nextflow options
runName : sleepy_mendel
containerEngine : singularity
launchDir : /Users/training/Downloads/test
workDir : /Users/training/Downloads/test/work
projectDir : /Users/training/Downloads/test/nf-core-demo
userName : training
profile : test,singularity
configFiles : /Users/training/Downloads/test/nf-core-demo/nextflow.config
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon/samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : my_results
Reference genome options
genome : R64-1-1
fasta : s3://ngi-igenomes/igenomes/Saccharomyces_cerevisiae/Ensembl/R64-1-1/Sequence/WholeGenomeFasta/genome.fa
Institutional config options
config_profile_name : Test profile
config_profile_description: Minimal test dataset to check pipeline function
Max job request options
max_cpus : 2
max_memory : 6.GB
max_time : 6.h
!! Only displaying parameters that differ from the pipeline defaults !!
------------------------------------------------------
If you use nf-core/demo for your analysis please cite:
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
------------------------------------------------------
executor > local (6)
[6e/df7039] process > NFCORE_DEMO:DEMO:INPUT_CHECK:SAMPLESHEET_CHECK (samplesheet_test_illumina_amplicon.csv) [100%] 1 of 1 ✔
[a6/cc842c] process > NFCORE_DEMO:DEMO:FASTQC (SAMPLE2_PE_T1) [100%] 4 of 4 ✔
[97/61b9c7] process > NFCORE_DEMO:DEMO:MULTIQC [100%] 1 of 1 ✔
-[nf-core/demo] Pipeline completed successfully-
1.3.3 Default nf-core configuration files
All of an nf-core pipeline's default parameters, configuration options, and profiles, are either defined in the pipeline's nextflow.config file, or are imported into that file from one of the .config files stored in the conf/ directory. The use of multiple .config files helps to logically separate different configuration options, such as the default and test configurations. Some of these .config files are included by default, while others can be optionally activated via profiles. The most important of these are:
base.config- Included by the pipeline by default
- Generous resource allocations using labels
- Does not specify any method for software management and expects software to be available (or specified elsewhere)
modules.config- Included by the pipeline by default
- Module-specific configuration options (both mandatory and optional)
- Note: This file isn't 100% consistent between pipelines. Some pipelines (e.g.
nf-core/sarek) have aconf/modules/folder with per-module.configfiles inside.
test.config- Only included if specified as a profile (
-profile test) - A configuration profile to test the pipeline with a small test dataset
- Only included if specified as a profile (
test_full.config- Only included if specified as a profile (
-profile test_full) - A configuration profile to test the pipeline with a full-size test dataset
- Only included if specified as a profile (
nf-core pipelines are also required to define software containers and conda environments that can be activated using profiles. Although it may be technically possible to run the pipelines with software installed by other methods (e.g., environment modules or manual installation), this is highly discouraged, since these pipelines have been developed and tested using containers and conda. Using containerisation systems like Docker or Singularity is also much more convenient and ensures reproducibility.
Tip
If you're computer has internet access and one of Conda, Singularity, or Docker installed, you should be able to run any nf-core pipeline with the test profile and the respective software management profile 'out of the box'. The test data profile will pull small test files directly from the nf-core/test-data GitHub repository and run it on your local system. The test profile is an important control to check the pipeline is working as expected and is a great way to trial a pipeline. Some pipelines have multiple test profiles for you to trial.
Shared configuration files
An includeConfig statement in the nextflow.config file is also used to include custom institutional profiles that have been submitted to the nf-core config repository. You can see a list of all current configs on the nf-core website. At run time, nf-core pipelines will fetch these configuration profiles from the nf-core config repository and make them available. They can be activated using the same -profile syntax discussed above.
For shared resources such as an HPC cluster, you may consider developing your own shared institutional profile. You can follow this tutorial for more help.
1.3.4 Pipeline parameters
Every nf-core pipeline defines a set of parameters that can be used to pass data, filtering thresholds, options, and other information to the pipeline and influence how it runs. These parameters are defined and given sensible defaults in the main nextflow.config file in the pipeline repository. While these default values are chosen to be applicable to the average user, you will almost certainly want to modify some of these to fit your own purposes and datasets.
IMPORTANT!
nf-core pipeline parameters must be passed via the command line (--<parameter>) or Nextflow -params-file option. Custom config files, including those provided by the -c option, can be used to provide any configuration except for parameters.
We discuss both of these methods for passing parameters further down.
To help with setting parameters, the full list of parameters each nf-core pipeline accepts is described on its page on the nf-core website. This parameter list includes a description of each parameter and the type of value it accepts. Most parameters will have additional text to help you understand when and how a parameter should be used. For example, the following is from the parameter page for nf-core/rnaseq:
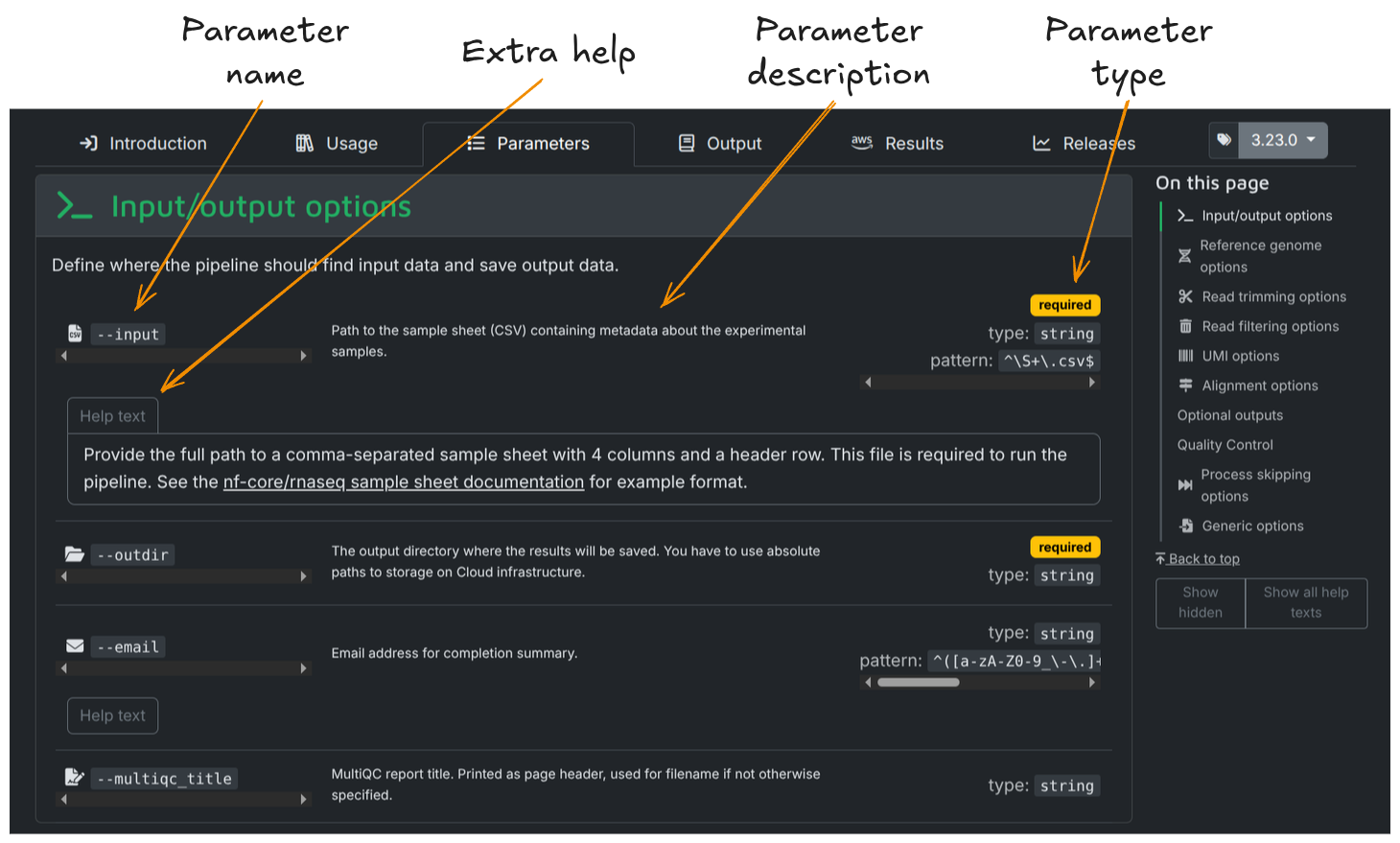
Parameters and their descriptions can also be viewed in the command line using the run command with the --help parameter:
Exercise 1.3.4 1 min
Use the --help parameter to view the available parameters for our Sydney-Informatics-Hub/nf-core-demo pipeline:
You should see:
N E X T F L O W ~ version 25.10.4
Launching `nf-core-demo/main.nf` [drunk_sammet] DSL2 - revision: 282d7ef11e
------------------------------------------------------
,--./,-.
___ __ __ __ ___ /,-._.--~'
|\ | |__ __ / ` / \ |__) |__ } {
| \| | \__, \__/ | \ |___ \`-._,-`-,
`._,._,'
nf-core/demo v1.0dev
------------------------------------------------------
Typical pipeline command:
nextflow run nf-core/demo --input samplesheet.csv --genome GRCh37 -profile docker
Input/output options
--input [string] Path to comma-separated file containing
information about the samples in the experiment.
--outdir [string] The output directory where the results will be saved.
You have to use absolute paths to storage on Cloud infrastructure.
--email [string] Email address for completion summary.
--multiqc_title [string] MultiQC report title. Printed as page header,
used for filename if not otherwise specified.
Reference genome options
--genome [string] Name of iGenomes reference.
--fasta [string] Path to FASTA genome file.
Generic options
--multiqc_methods_description [string] Custom MultiQC yaml file containing
HTML including a methods description.
FastQC options
--kmer [integer] FastQC kmer length [default: 7]
!! Hiding 24 params, use --show_hidden_params to show them !!
------------------------------------------------------
If you use nf-core/demo for your analysis please cite:
* The nf-core framework
https://doi.org/10.1038/s41587-020-0439-x
* Software dependencies
https://github.com/nf-core/demo/blob/master/CITATIONS.md
------------------------------------------------------
1.3.5 Setting pipeline parameters on the command line
One way of setting pipeline parameters is directly via the command line. Any parameter can be configured on the command line by prefixing the parameter name with a double dash (--):
Parameters set via the command line have the highest precedence and will override conflicting parameters defined anywhere else.
Tip
Remember that pipeline parameters are prefixed with a double dash (--), while options to the nextflow run command itself are prefixed with a single dash (-).
All parameters take arguments, i.e. a value that they get set to. Arguments are specified on the command line immediately after the parameter name:
Note that string arguments must be wrapped in quotes if they contain spaces:
Some parameters are boolean, i.e. they are set to either true or false. These are typically used to turn some pipeline functionality on or off. To set these, you simply write true or false after the parameter name:
A special case is when you are setting a parameter to true. In this case, you can omit the word true:
nextflow run <pipeline> --some_boolean_parameter
# Equivalent to:
nextflow run <pipeline> --some_boolean_parameter true
Exercise 1.3.5 3 mins
The nf-core-demo pipeline generates a MultiQC report. From the help information we printed out before, we can see that one of the pipeline parameters can be used to customise the title of this report. Run the nf-core-demo pipeline and use this parameter to customise the title of the MultiQC report with the name of your favourite animal.
Add the --multiqc_title flag to your nextflow run command and provide it the name you chose. If you chose a name containing spaces, ensure you wrap the string in double quotes first. Don't forget to provide the test and singularity profiles as well! Additionally, use the -resume flag to avoid re-running the entire pipeline again.
In this example, you can check your parameter has been applied by listing the files created in the results folder (my_results):
--multiqc_title is a parameter that directly impacts a result file. You should see something like the following:
For parameters that are not as obvious, you may need to check your log to ensure your changes have been applied. You can not rely on the changes to parameters printed to the command line when you execute your run:
```console title="Output" hl_lines="6" ...
NFCORE_DEMO:DEMO:MULTIQC
multiqc \
--force \
--title "koala" \
--config multiqc_config.yml \
\
.
cat <<-END_VERSIONS > versions.yml
"NFCORE_DEMO:DEMO:MULTIQC":
multiqc: $( multiqc --version | sed -e "s/multiqc, version //g" )
END_VERSIONS
...
```
1.3.6 Using parameter files
Setting parameters via the command line might be ok for one or two parameters, but for a typical nf-core pipeline you may need to customise a lot of parameters to suit your use case. Using the command line to set lots of parameters like this can be error-prone and messy, so Nextflow offers another method: parameter files.
Parameter files are either JSON (.json) or YAML (.yaml or .yml) files that can contain an unlimited number of parameters as key-value pairs:
{
"parameter1_name": 1,
"parameter2_name": "a string",
"parameter3_name": true
}
parameter1_name: 1
parameter2_name: "a string"
parameter3_name: true
The above examples are equivalent to passing --parameter1_name 1 --parameter2_name "a string" --parameter3_name true on the command line.
You can override a pipeline's default parameters by creating a custom parameter file like this and passing it as a command-line argument using the -params-file option.
This keeps all of your parameters together in a separate file and keeps your nextflow run command nice and clean.
Exercise 1.3.6.1 5 mins
Run the nf-core-demo pipeline again and use a custom parameter file to set the title of the MultiQC report to the name of your favourite food.
Create a file called my_custom_params.json that contains your favourite food, e.g., cheese:
Include the custom .json file in your execution command with the -params-file option:
Check that it has been applied:
Remember that the parameters specified on the command line, e.g. --paremter_name, will take precedence over any conflicting values specified in a parameter file with -params-file.
Exercise 1.3.6.2 2 mins
Run the nf-core-demo pipeline again using the custom parameter file you made in Exercise 1.3.6.1. In addition, use the --<parameter_name> syntax to set the title of the MultiQC report to the name of your favourite colour.
Solution
Use the same command that you used in Exercise 1.3.6.1, but this time add --multiqc_title along with your favourite colour:
nextflow run nf-core-demo -profile test,singularity -params-file my_custom_params.json --multiqc_title blue -resume
Check the results to see which one was applied:
The value supplied to --multiqc_title on the command line (blue) took precedence over the value supplied in my_custom_params.json (cheese). Remember, parameters specified on the command line take precedence over parameters set in -params-file or .config files.
1.3.7 Custom configuration files
Nextflow can load additional, custom configuration options from any arbitrary .config file supplied to the -c option on the command line:
This allows you to customise the configuration of the Nextflow run beyond the defaults and what is set by any institutional profile. Furthermore, multiple custom .config files can be included at execution by separating them with a comma (,). Note that the order matters, with the files being read in left to right, and the right-most files taking precedence.
Custom configuration files follow the same structure as the main nextflow.config file included in the pipeline directory. Configuration properties are organized into scopes by dot-prefixing the property names with a scope identifier or grouping the properties in the same scope using the curly brackets notation. For example:
is equivalent to:
Scopes allow you to quickly configure settings required to deploy a pipeline on different infrastructure using different software management. For example, the executor scope can be used to provide settings for the deployment of a pipeline on a HPC cluster. Similarly, the singularity scope controls how Singularity containers are executed by Nextflow. Multiple scopes can be included in the same .config file using a mix of dot prefixes and curly brackets. See the Nextflow documentation for more information on writing configuration files, as well as this list of configuration options.
Exercise 1.3.7 3 mins
Run the nf-core-demo pipeline again and use a custom config file to set the title of the MultiQC report to the name of your favourite month.
Create a file called my_custom_config.config and set the multiqc_title parameter within the params scope to your favourite month, e.g., October:
Alternatively, use the longer scope block format:
Include the custom .config file in your execution command with the -c option:
What happened?
Check that it has been applied:
It didn't work!
This is confusing, because the custom value we supplied for the multiqc_title parameter is listed in the terminal output:
...
Input/output options
input : https://raw.githubusercontent.com/nf-core/test-datasets/viralrecon samplesheet/samplesheet_test_illumina_amplicon.csv
outdir : my_results
multiqc_title : October
...
However, it was not actually applied during the execution of the command:
...
NFCORE_DEMO:DEMO:MULTIQC
multiqc \
--force \
\
--config multiqc_config.yml \
\
.
cat <<-END_VERSIONS > versions.yml
"NFCORE_DEMO:DEMO:MULTIQC":
multiqc: $( multiqc --version | sed -e "s/multiqc, version //g" )
END_VERSIONS
...
Why did this fail?
When using nf-core pipelines, you cannot use the params scope in custom configuration files. Parameters can only be configured using either a parameter file and the -params-file option, or using the --<parameter_name> syntax directly on the command line.
1.3.8 Configuring processes
Within a .config file, the process scope allows you to configure workflow processes and is used extensively to define resources and additional arguments for modules.
By default, process resources for nf-core pipelines are allocated in the conf/base.config file using the withLabel selector:
The above configuration will target any process given the label BIG_JOB and set its required CPU count to 16 and its required memory allocation to 64 GB.
Similarly, the withName selector enables the configuration of a process by name. By default, per-module configurations are defined in the conf/modules.config file.
The above configuration will specifically target the MYPROCESS process and set its required CPU count to 4 and its memory allocation to 8 GB.
Tip
Note that withName selectors take precedence over withLabel selector. In the above example, if the process MYPROCESS also had the label BIG_JOB, it would only be assigned 4 CPUs and 8 GB of memory as per the withName configuration.
Most tools have lots of parameters that can be set on the command line. A pipeline might expose the most important and frequently used of these as pipeline parameters that can be directly configured via the --<parameter_name> syntax or in a parameter file.
However, it often isn't feasible to handle all of the tool's parameters. Instead, most nf-core pipelines define an ext.args process directive in the conf/modules.conf file. This is used to pass arbitrary command line arguments through to the underlying tool and helps to keep modules sharable across pipelines.
In fact, we've already seen this in action. The pipeline parameter --multiqc_title we configured earlier is passed to the ext.args directive of the MULTIQC process in conf/modules.config:
...
process {
...
withName: MULTIQC {
ext.args = params.multiqc_title ? "--title \"$params.multiqc_title\"" : ''
}
...
}
...
The value of ext.args then gets passed directly to the multiqc tool verbatim. You can use this too in your custom configuraiton files: if you wanted to add a specific argument to multiqc that wasn't currently supported by the nf-core-demo pipeline, you could use the process scope in your custom configuration file:
However, if a process is used multiple times in the same workflow, an extended execution path of the module may be required to make it more specific:
The extended execution path is built from the workflows, subworkflows, and modules used to execute the process.
In the example above, the nf-core MULTIQC module was called by the DEMO workflow, which was called by the NFCORE_DEMO workflow in the main.nf file.
Tip
It can be tricky to evaluate the extended path used to execute a module. If you are unsure of how to build the path you can use nextflow log to print the full process names:
Exercise 1.3.8 5 mins
Use a custom config file to set the ext.args directive for MULTIQC:
- Open
my_custom_config.configagain and remove the failedparamsscope - In its place, add a
processscope - Use a
withNameselector to specifically target theMULTIQCprocess - Use
ext.argsto set the--titleparameter of themultiqctool to your favourite month of the year, e.g,'--title "october"':
process {
withName: "NFCORE_DEMO:DEMO:MULTIQC" {
ext.args = '--title "october"'
}
}
Execute your run command again with the custom configuration file:
Check that it has been applied:
We can also look at the log output to see the extra arguments being applied:
1.3.9 Custom profiles
In some cases, you may want to be able to turn an entire set of parameters or process directives on or off at the same time. In these cases, you might prefer to write a custom profile to group all of these settings together and turn them on with the -profile syntax.
Profiles can be defined in a .config file using the profiles scope:
Within the profile definition, you can include any of the normal configuration options, such as process directives and parameters. You can also use the includeConfig option within a profile definition to include an entire .config file whenever you use the profile:
Exercise 1.3.9 5 mins
Create a custom profile that includes the config file we built earlier in Exercise 1.3.8.
Before trying this exercise, clean up the existing working directory by deleting the my_results directory.
Now, create the new custom profile:
- Create a new file called
profiles.config - Add a
profilesscope - Create a new profile called
myprofile - Add an
includeConfigstatement to the new profile and include themy_custom_config.configfile from Exercise 1.3.8. - Try running with and without the new profile enabled.
Run the pipeline, using -c to include the new profiles.config file:
You should notice that the MultiQC report has returned to its default title:
Run the pipeline again, but supply the new profile by adding myprofile to the -profile option:
Now you should see that the MultiQC report has the custom title again:
Key points
- nf-core pipelines follow a similar structure
- nf-core pipelines are configured using multiple configuration sources
- Configuration sources are ranked to decide which settings to apply
- Pipeline parameters must be passed via the command line (
--<parameter>) or Nextflow-params-fileoption